21 CFR Part 11の経緯

21 CFR Part 11の経緯
21 CFR Part 11について研究するページです。
*万が一文中に解釈の間違い等がありましても、当社では責任をとりかねます。
本文書の改訂は予告なく行われることがあります。
21 CFR Part 11の経緯
FDAは、1997年8月20日に21 CFR Part 11 ”Electronic Records; Electronic Signatures”(以下、Part11)という規則を発行した。
FDAに限らず、米国国防総省、財務省、環境保護庁では、「機密性の高い電子業務の実現には、安全性が保証される必要がある」との認識を持っている。
Part11は、米国の規則の中で最も早く発表されたペーパレスに関する規制要件である。その後、多くの法律や規則の立案の参考にされたとされる。
たとえば、1998年には「Office of Management and Budget」(政府事務合理化法)が制定された。
また2000年12月末、行政管理予算局(OMB)から発令された政府機関のペーパーワークを除去するための法律「U.S. Government Paperwork Elimination Act」(GPEA:政府書類作業排除法)が出されている。これは、2003年までにサービスや書式をオンラインで利用可能にして書類の提出も電子的に行えるようにすると規定されている。
実は、Part11をはじめペーパレスに関する規則等は”規制要件”というよりも、これまで紙でしか認めなかった記録や署名を、電子で認めるという”規制緩和”であるということである。
またPart11は、電子記録を義務付けるものではない。これまでどおり、紙の記録は使い続けても良いのである。
既存システムのPart 11対応(すなわち再設計、再バリデーション)はすぐには実施が困難である。
しかしながら、Part 11の要求事項の中には、運用により対応できる事項も多い。
Part11の目的は以下のようなものである。
またその骨子は、以下のようなものである。
- オーディットトレール(監査証跡)
・誰が、いつ、何をしたのかの完全な記録をとる
・変更前・変更後の完全なログをとる - セキュリティ・インテグリティ(安全性・完全性)
・アクセス・コントロールを行う
・データを、上書き・変更・削除等から保護する - バリデーション
・コンピューターシステムやソフトウェアのバリデーションを行う
・バリデーションの記録を作成する
特にセキュリティと監査証跡の2つの要件は重要である。
この2つの要件は、たとえSOX法やPart11や厚労省ER/ES指針が、何回改定されようとも削除されることのないものである。
もしセキュリティか監査証跡のどちらか一方の機能を持たないシステムである場合、その電子記録は信用することができないからである。
CFRとは
CFRは、Code of Federal Regulations(連邦行政規則集)の略であり、米国には50ものCFRが存在する。
CFRの第1巻は「President」であり、第2巻は「Financial」である。
21巻が「Food and Drug」であり、FDAが受け持っている。
また21 CFRにはPart(章)が1から1,499まで用意されており、それぞれカテゴリによって区別されている。
CFR制定のステップ
米国では、政府が何を考え、何をしようとしているのかを、常に国民が知る権利を持っている。
そのため、FDAは「規則(regulation)」の立案等に際して、連邦広報(Federal Register:FR)という日本における「官報」に相当するものを用いて、広く国民に知らせなければならない義務がある。
規制当局が規則を施行させるまでには、以下のような段階を踏まなければならない。
第一段階:「規則制定の事前通告」を行う
第二段階:「規則案の通告」とパブリックコメントの募集を行う
最終段階:「最終規則の公示」とパブリックコメントに対する回答、規則の施行日を通告
21 CFR Part 11を例にとってみると、1992年に第一段階である「規則制定の事前通告」がFRに掲載された。
その後、第二段階として1994年8月31日のFRに「21 CFR Part11電子記録・電子署名」の規則案(Draft Rule)を発表した。同時に90日間、パブリックコメントの募集を行った。規則案をFRに公示しても、案に対する業界からの反対が多くて最終規則にまで至らないか、ボツになるか、規則がいつまでも実現しないことがよくある。
最終段階である「最終規則の公示」では、「21 CFR Part11電子記録・電子署名」の最終規則(Final Rule)が1997年3月27日のFRに掲載された。
この最終規則の発表時には、規制当局がパブリックコメントをどう考え、規則案をどのように修正したかをpreambleと呼ばれる前文に記載しなければならない。
Part 11の場合は、条文本体が3ページ弱なのに比べ、このpreambleが35ページにも及ぶ。
またこの最終段階では同時に規則の施行日を指定しなければならない。Part 11は、1997年8月20日がその施行日と指定された。
このように米国では、政府の規則立案にあたっては、透明性を求められる。
ガイダンス
CFR以外にも、FDAはGuidanceを発行することがある。GuidanceはCFRの下位に位置し、業界向けガイダンス(Guidance for Industry)とFDA職員向けガイダンス(Guidance for Staff)の2通りがある。Guidanceの内容はあくまでもFDAの推奨事項であり、代替方法の採用は構わないと記載されていることが多い。
ガイダンスには、業界向け(Guidance for Industry)とFDAスタッフ向け(Guidance for Staff)またはその両者向けがある。
Part11発行後には、関連するガイダンスとしてGlossary of Terms、 Validation、Time Stamps、 Maintenance of Electronic Recordsなどのガイダンスが発行された。
またPart11の改定を示唆した「Guidance for Industry Part11, Electronic Records; Electronic Signatures – Scope and Application」は重要である。
21 CFR Part 11とは
Part11は、システムが一定の基準を満たせば、電子記録、電子署名を従来の紙での記録および手書き署名に相当すると考える判断基準であり、電子申請への対応を可能にするための規則である。(図5参照)

図5.21 CFR Part 11とは
FDAは、Part11の発行に際してこう述べている。
「Part11はこの20年で最も重要なFDAの規則である。業界が規則の要求事項に準拠することで技術的に前進していくことを可能にしている。Part11は「情報化時代」においてデータの完全性を維持するための「Common Sense」アプローチである。」
しかしながら、Part11を読むにつけ、実際にはその内容は、いかに不正を防止し、不正を発見するかに終始しているように思われる。
けっして、業界の技術的な進歩を支援するようなものにはなっていないのである。
Part11の歴史
それでは、Part11制定の歴史と、制定後の動向について時系列に解説しよう。まずそれらの年表を図6に示す。
| 1990 年代初頭 | 電子署名(ペーパレスシステム)への認識始まる |
| 1990 | 米国製薬工業協会がFDAに電子署名使用についての指針を示すよう申入れ |
| 1992 | 規制立案の事前通告 − FDAとしての見解の概念、業界との話し合い |
| 1994.8.31 | 「 21 CFR Part11電子記録・電子署名」規制案(Draft Rule)公示 |
| 1994 |
|
| 1997.3.20 | 「21 CFR Part11」(Final Rule)発行 |
| 1997.8.20 | 「21 CFR Part11」施行 「The final rule on electronic records、signatures、and submissions」 |
| 1999 | FDAが業界トレーニングを実施 |
| 1999.4 | Computerized System Used in Clinical Trials |
| 1999.5 | Compliance Policy Guide 7153.17公表 |
Partt11に対する産業界からの緩和要請 |
|
| 2003.2.4 | 「電子記録の電子コピー」取り下げ cGMP指導要領と矛盾 |
| 2003.2.20 | Guidance for Industry Part11 ER;ES – Scope and Application (Draft)公表 |
| 2003.9 | Guidance for Industry Part11 ER; ES – Scope and Application(Final)発行 |
| 2004.6.11 | Public Meeting 開催中止 |
| 2004.9.29 | 21世紀 GMPイニシアティブの最終報告発行 「21 CFR Part 11の改定案を2005年中に発行する。」 |
| 2010.7.8 | 人体用医薬品に関し21 CFR 11 (Part 11) 要件に焦点を合わせた査察の実施を発表 |
図6.Part11の歴史
Part11の立案と施行
1990年代の初頭に、米国の製薬業界でペーパーレスに関する関心が高まった。
なぜならば、医薬品の製造における製造記録などは、目まぐるしく電子化が進んだのであるが、最終的に手書きによる署名を行わなければならなかったため、結果的にすべての記録を印刷せざるを得なかったからである。もし規制当局が電子署名を認めるならば、完全なペーパーレスが実現でき、効率化が図れることになる。
1991年には、米国製薬工業協会 (当時PMA、現PhRMA)がFDAに対し、電子署名の使用についての指針を示すように申し入れた。
これを受けて、FDAは多部門にまたがるタスクフォース(a Task Force on Electronic Identification/Signature :電子認証・電子署名に関するタスクフォース)を組織した。
1991年には、Paul J.Motiseを責任者として、検討のための作業部会を設け、現行のGxP規則のもとで、どのようにすればペーパーレスシステムが実現できるかについて、業界と打合せを重ねた。
1992年には、CFR制定のための第一段階である、規制立案の事前通告”Advanced Notice of Proposed Rulemaking”を連邦広報に掲載し、意見を求めることとした。
これは従来の紙の記録や手書き署名に代えて、新しい技術を導入する際に解決すべき重要な問題は何かについて、FDAの見解を示したものであった。
FDAには数多くのコメントが寄せられ、その後、2年間にわたり業界との話し合いが続けられた。
そして1994年8月31日の連邦広報で、第二ステップである、規則案(Draft Rule)の公示を行った。
この規則案に対し、広くパブリックコメントを募集し、90日以内にコメントを出すよう要請した。
規則案は、業界の予想したものとは大きくかけ離れ、実現が困難なものや、適合するためには多大なコストや労力の負担を強いるものであった。
これに対し、医薬品業界から49のコメントが提出された。この49のコメントには、回答が困難なものが多く含まれていた。
FDAは、コメントに応えて、負担を軽くするよう改定作業を行った。
この改定作業は困難なものであったと推察される。なぜならば最終規則が公示されるまで3年弱を必要としたからである。
またパブリックコメントの回答にあたるpreumble(前文)は、Part11本体が連邦広報に掲載された際にたった2ページ半であるのに対して、35ページも及ぶものとなったのである。
ともあれ、Part11は、1997年3月20日の連邦広報で公示され、5ヶ月後の8月20日をもって施行することとなった。
Part11施行後の動向
1999年には、FDAが業界トレーニングを実施した。
1999年4月には「Computerized System Used in Clinical Trials」と呼ばれるガイダンスが発行された。
このガイダンスは、臨床試験の現場(すなわち医療機関)においてコンピュータ化システムを利用する際の要件を記載したものである。Part11発行以降に出されたガイダンスであり、FDAのPart11に対する考え方を具体的に理解することができるものである。なおこのガイダンスは、2007年5月11日に改定されている。
Part11は、立案の歴史から、電子署名に重きを置いた規則となっている。しかしながら、運用し、理解を深めていくうちに、電子署名よりも電子記録が重要であることにFDAも業界も認識するようになってきた。
Compliance Policy Guide 7153.17
1999年5月13日には、「Compliance Policy Guide 7153.17」(以下、CPG 7153.17)を公表した。このCPG 7153.17は、Part 11に対するFDAの考え方を示したものであり、査察や是正措置執行に際する手引きとなっている。(図7参照)

図7.CPG 7153.17
CPG 7153.17のキーコンセプトは “正確性、信頼性、意図した性能の一貫した確保、ならびに無効となったり変更されたりした記録を、識別する能力が保証されるようにするためのシステムのバリデーション”としている。
FDAは電子記録の一貫性・正確性(Integrity)について関心があり、
ということを要求しているのである。
具体的にはコンピュータ中のデータは、セキュリティで保護されており、なおかつ変更をはじめ操作内容を自動的に記録する監査証跡機能(Audit Trail)を持たなければならないということである。
実際Part11施行以来、セキュリティ、監査証跡、電子記録の一貫性・正確性に関する指摘が増加している。ただしCPGでは、FDAの査察ではGxP対応の観点で判断し、Part 11のみでの査察・判断はしないとしている。(図8参照)
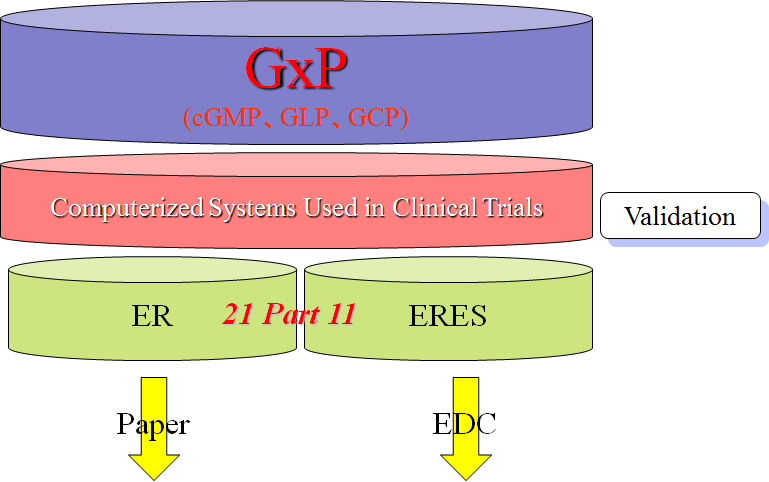
図8.GxP対応の観点で判断
Part11に関するFDAガイダンスとその後の動向
Part11には、条文解釈の難解さや機能・運用両面での多くの適合要件と実現の困難さといった問題があった。
Part11の条文 は、たったの3ページに過ぎないため、業界はより詳しいガイダンスの発行を要求した。
このためFDAは、以下の6つのガイダンスを発行することを約束した。
- バリデーション(Validation)
Part11対応の前提としてのCSVの取り組み指針 - 電子記録の維持管理(Maintenance of Electronic Records)
電子記録を長期保存する場合の課題とその対応指針 - 電子記録のコピー (Electronic Copies of Electronic Records)
査察時などに規制当局等へコピーを提出する場合の指針 - タイムスタンプ(Time Stamps)
監査証跡に含まれる操作時間の考え方や、システム聞の時刻合わせなどに関する指針 - 用語の定義(Glossary of Terms)
解釈が分かれる用語などの定義 - 監査証跡(Audit Trails)
未発表
これらのガイダンスは、監査証跡を除き、産業界からのコメント収集を目的としたドラフト(草案)として順次発行され、Part11への適合作業を進める関係者にとって重要な指針であった。
最終的に監査証跡を除く、5つのガイダンスが発行されたが、より厳しい要求となってしまった。
その後FDAは、これらすべてのガイダンスを取り下げるという前代未聞の発表を行った。
Part11に関する業界の動向
Part11の適用要件を精査していくと、それは電子記録・電子署名にとどまらず、電子化要件の基盤であるコンピュータ・システム・バリデーション(以下、CSV)や、操作履歴の保存をはじめとする運用管理も合まれていることが明らかになってきた。
しかもPart11の適用範囲は医薬品の製造・品質規範であるGMP(Good Manufacturing Practice)にとどまらず、FDA規制対象の全体を包含した、広範囲にわたる規制ともなっており、このため製薬企業では、生産部門だけでなく研究開発(GLP)、臨床開発 (GCP)、IT(情報技術)、品質保証など、多くの部門がPart11にかかわることが判明した。
さらにPart11は、既存システム(レガシーシステム)に関しでも、免除は一切認めなかった。
すなわち、Part11は、施行時点で稼動中のシステムも対象とするという厳しい規制内容であり、そのことから、大手製薬企業のなかには数千、中堅の製薬企業でも数百の既存システムへの適合作業を実施することになり、相当な作業量と時間、そして費用が必要となることもわかってきた。
このような広範囲な対応を行うために、企業側は莫大な費用と、長期間に渡る対応の必要性を懸念した。
また遵守要件が具体化していないため、対応方針、新システムへの移行に不安があった。
Part11が施行された後も、 FDAと産業界と規則の解釈や実行に関する議論やPart11に潜在する問題点のヒアリングを重ね、その過程で産業界からは、 Part11に適合させるための費用についての懸念が表明されていた。
たとえば米国研究製薬工業協会 (PhRMA)からは、加盟企業のPartl1対応費用は21億ドル(約2,500億円)を超えるという報告があり、日本国内からも、大手製薬企業のなかには 157億円の予算を立てた」という話も伝わってきていた。
また「某製薬企業は新薬の工場建設に当たり、当初の計画では完全自動化を行う予定であったが、Part11への対応が困難なため多くの従業員を採用し、手動の工場にした」といった話もあった。
Scope and Application
米国製薬工業協会(PhRMA)の調査によると、Part11対応費用は21億ドルという結果であった。
Part11に対する産業界からの緩和要請が出され、「Part11に関する業界連合」が結成された。
FDAは、cGMP指導要領と矛盾するという理由から、2003年2月4日に「電子記録の電子コピー」を取り下げた。
こうしたこともあって、 FDAはPart11の規則の再検討に乗り出し、2003年9月には「Guidance for Industry Part11, Electronic Records; Electronic Signatures – Scope and Application」(以下、Scope and Application)と呼ばれるガイダンスを発表した。このガイダンスは、Part 11のスコープと適用に関するFDAの最近の考え方を述べたものである。
Part11再検討の理由は「Scope and Application」のなかで明らかにされた。
すなわち、 Part11の条文に対して厳しい解釈がなされる方向であったことが、ドラフトルールを作成した時点では予期されなかったほど適合コストを増加させ、革新と技術の進歩を阻害したからであるとされた。
そこでScope and Applicationで、はPartl1の適用範囲を狭義に解釈し、Part11の重要な要件で、ある監査証跡、記録の保存、既存システムの適用などは一定の条件を示したうえでの施行裁量として、規制の対象としないこととした。
すなわち、過度になりすぎた一部のガイダンスや、 FDAの一部職員による過大な解釈による行きすぎた要求が、あたかもFDA全体の考え方であるような誤解を与えてきたという反省が、Scope and Applicationの発行になったとも考えられる。
それだけではなく、Scope and Applicationの発行にはもう一つ伏線があった。
FDAは前年の 2002年 8月に 「pharmaceutical cGMPs for the 21st Century : A Risk-Based Approach」を発表し、 21世紀の「規制に対する取り組み」に当たって、リスクベースに基づいた科学的な指導をするという新しいアプローチを宣言していた。つまり「範囲と適用」は、これを受けての「リスクベースアプローチのPart11」だったのである。
このガイダンスの中でFDAは、Part11を見直すことを宣言した。その理由として、
- 当初の意図にそぐわない方法による不要な制限
- コンプライアンスコストの著しい増加
- 公衆の健康に利益を与えず、技術革新を阻害
を挙げている。
そして、従来発行してきたドラフトガイダンスのすべてと、CPGの取り下げを発表した。
- CPG 7153.17 - Compliance Policy Guide
- Glossary of Terms
- Validation
- Time Stamps
- Maintenance of Electronic Records
このガイダンスによりFDAは以下を推奨している。
- Part 11を狭く解釈し、Part 11の対象となる電子記録を少なくする。
- 「GxP記録」を定義し、Risk Based Approachをとる。すなわち「電子記録とは何か」というボトムアップアプローチではなく、「GxP記録か」というトップダウンアプローチ(Risk Base Approach)をとる。
- 執行を裁量する。(レガシーシステムなど)
- Part 11の対象となる記録については、プレディケート・ルールの要件を満たすこと。
- バリデーションへの適合(General Principles of Software ValidationやGAMP 4を参考に)
注意しなければならないことは、「21 CFR Part11」および「Scope and Application Guidance」は、今もって有効であるということである。
パブリックミーティングの中止
FDAが2004年6月11日に開催を予定していたPart11 Public Meetingが中止となった。
中止の理由は、故レーガン元大統領の国葬により政府機関が臨時休暇とされたためであった。
しかしながら、再開催を行わなかったことからもわかるように、実際には開催したとしても更に混乱を増すばかりであったものと推察できる。
Part11を改定し、本格的に実施したいFDAに対して、全面的にPart11廃止を要求する業界側とでは、全く意見が対立している。
このままでは、公開討論を行っても収集がつかないのは明らかだったであろう。
いったいFDAの意図はどこにあるのか、またPublic Meetingに何を求めて、どこにゴールをおいていたのかが不明である。
21 CFR Part 11の査察開始(再開)
筆者が知る限り、Part11によるワーニングレターが出されたのは、2001年10月が最後である。
これは、これまでに述べたとおりの、FDAと業界との軋轢により、事実上Part11査察が止められてきたからである。
しかしながら、2010年7月8日、FDAは突如、ヒト用医薬品に関して、21 CFR Part 11の査察開始(再開)をアナウンスした。
これはPart11の改定を助けるもので、業界がどれだけPart11を理解しているのかを調査するものだという。
なぜこの時期に再開されたかというと、FDA担当官が代わったこと。また最近のGMP査察において、データの完全性が損なわれていた事例が発見されたからである。
本アナウンスによる査察は、技術に着目しているのではなく、データの完全性に着目している。Part11についての原理、原則はなんら変わっていない。技術がどう変わろうとPart11の重要性に変わりはない。
したがって、電子記録の原理原則と要求を満たしていれば良いのである。
以下は、FDAはPart11査察を再開するとしたアナウンスの全文である。
当局(FDA)は一連の査察を実施する予定である。 電子記録、電子署名、および電子記録に実施された手書き署名は、信頼性、信用性があり、紙の記録と紙に手書きされた署名と同等であると当局はみなしているが、そのみなす基準を説明する規則が21 CFR 11(Part 11)である。 背景 |


