
1. �͂��߂�
�����J���Ȃ́A2010�N7��16���u���i�E��O�i�����̔��Ǝғ��ɂ�����R��
�s���[�^���V�X�e���K���Ǘ��K�C�h���C���i�āj�v�\���A�p�u���b�N�R�����g�̕�W���J�n�����B
�p�u���b�N�R�����g�́A8��20���܂Ŏt���Ă���B
�܂��V�K�C�h���C���i�āj�ɉ����āA�l�����iQ&A�W�j�������Ɍf�ڂ���Ă���B
���̃K�C�h���C���́A�u�R���s���[�^�g�p���i���������K���Ǘ��K�C�h���C���v�i�����S�N�Q���Q�P����đ�11���F����17�N3��30���t��H�Ė�����0330001���ɂ��p�~�j��u����������̂ł���B
���K�C�h���C���́A���Ă̋K���v���ɑ��āA��r�I���e����̓I�ŗ������₷���A
���p�I�ł������B
�������Ȃ���AIQ�AOQ�APQ�Ƃ��������i�o���f�[�V�����j�Ɋւ���L�q���Ȃ��A
�O���̋K�����ǂ̍��@�����ɋꗶ����Ƃ�������ʂ��������B
����͌��J�ȔŁu�R���s���[�^���V�X�e���K���Ǘ��K�C�h���C���i�āj�v���l�@���Ă݂����B
2.�@�V�K�C�h���C���̓���
�V�K�C�h���C���i�āj����ʂ�ǂ�ۂł́A�ȉ��̓���������悤���B
1) GMP�AGQP �����ΏۂƂ��Ă���B
2) ���K�C�h���C����GAMP 5 ��ܒ������悤�ȓ��e�ł�
��B
3) FDA ��ANNEX11 ���̗v�������ɔ�ׂāA��̓I�ł���B
4) �R���s���[�^���V�X�e���̊J������A���A�^�p�Ǘ��y�єp���܂ł����C�t�T�C�N���Ƃ��Ē�`���Ă���B
5) �����̗p�ꂪ���{��ŕ\�L����Ă���B
6) �\�t�g�E�F�A�̃J�e�S�����ނ��`���Ă���B
7) DQ�AIQ�AOQ�APQ �Ƃ����p����g�p���Ă���B
8)�u �o���f�[�V�����v�Ƃ����p��̒�`�����̂��Ƃ��Ӗ����A���`�ł���B
2.1�@GMP�AGQP �����ΏۂƂ��Ă���
�u2. �K�p�͈̔́v�ɂ́A�u���̃K�C�h���C���́A�f�p�o�ȗߋy�тf�l�o�ȗ߂��K�p�����Ɩ��ɁA�R���s���[�^���V�X�e�����g�p���鐻���̔��Ǝғ��ɓK�p����B�v�Ƃ̋L�ڂ�����B
�������Ȃ���A���Ăł����l�ł���悤�ɁA��Տ�������Տ������Ȃǂ̌����J���ɂ����Ă��K�p�����̂ł͂Ȃ����ƍl������B
�Ȃ��Ȃ�AGLP ��GCP �ɂ����Ă��o���f�[�V�������v������Ă��邪�A���̍ۂɋ��菊�Ƃ���K���v���Ƃ��ẮA�{�K�C�h���C�����B��ł��邩��ł���B
2.2�@���K�C�h���C����GAMP 5 ��ܒ������悤�ȓ��e�ł���
�V�K�C�h���C���́AGAMP 4 ��GAMP 5 ���Q�l�ɂ��Ă���B
�K���v���ɋƊE�W�������p���邱�Ƃ́A�ٗ�ł���B
2.3�@FDA ��ANNEX11 ���̗v�������ɔ�ׂāA��̓I�ł���
�e���ʕ��̃R���e���c����`���Ă���B
�Ⴆ�ΐv�d�l���̓��e�ɂ����y���Ă��邪�A�{���v�d�l�����́A���Y�����҂�QMS �ɏ]���č쐬�����ׂ��ł���B
2.4�@�R���s���[�^���V�X�e���̊J������A���A�^�p�Ǘ��y�єp���܂ł����C�t�T�C�N���Ƃ��Ē�`���Ă���
���K�C�h���C���ł́A���C�t�T�C�N���Ƃ�����`�͂Ȃ������B�܂��V�K�C�h���C���ł́A�R���s���[�^�V�X�e���̔p���܂Ŕ͈͂Ƃ��Ă��邱�Ƃ������I�ł���B
2.5�@�����̗p�ꂪ���{��ŕ\�L����Ă���
�^�C�g�����u�K�C�h���C���v�ł���ɂ�������炸�A���Ɂu�����ҁv�u�J���ӔC�ҁv�u���ؐӔC�ҁv�Ȃǂ̓��{��\�L���݂���B
�����҂́A�T�v���C���̂��Ƃł���A�J���ӔC�҂͈�ʂɃv���W�F�N�g�}�l�[�W�����w���Ǝv����B�܂����ؐӔC�҂̓o���f�[�V�����}�l�[�W���̂��Ƃł���ƍl������B
����ɂ����āA�u�����҃A�Z�X�����g�v�Ȃǂ̃J�^�J�i�\�L���݂���B
2.6�@�\�t�g�E�F�A�̃J�e�S�����ނ��`���Ă���
�\�t�g�E�F�A�̃J�e�S�����ނ́AGAMP �ɓ����I�ȍl�����ł���B�J�e�S�����ނ́A���X�N�]���̈�ł���A�������傫���Ȃ�قǕύX�̓x�����������A�s����̃��X�N�����傷��B
2.7�@DQ�AIQ�AOQ�APQ �Ƃ����p����g�p���Ă���
���K�C�h���C���ɂ́A���؋Ɩ��Ɋւ���L�q���Ȃ��A����������DQ�AIQ�AOQ�APQ �Ƃ����p����L�q���Ȃ������B
�V�K�C�h���C���́AGAMP 4 ���Q�l�Ƃ��A�����̗p���p���Ă���B
�������Ȃ���AFDA ��GAMP 5 �ł́ADQ�AIQ�AOQ�APQ�Ƃ����p��͎g�p����Ȃ��Ȃ����B
�Ȃ��Ȃ�����̗p��͐���ƊE�ɓ����I�ȌĂѕ��ł���A��ʂɃT�v���C���ɂ͗�������ɂ������̂ł��邩��ł���B
�V�K�C�h���C���́A���ꂩ�甭�o�����ɂ�������炸�A����DQ�AIQ�AOQ�APQ �Ƃ����p����g�p���Ă���B���̏ꍇ�A���łɐ����Ђ�GAMP 5 ���̃O���[�o���X�^���_�[�h�ɑΉ�����CSV SOP���쐬���Ă����ꍇ�A�s�������N�����˂Ȃ��B
������_�u���X�^���_�[�h�̖�肪�����邱�Ƃ����O�����B
2.8�@�u�o���f�[�V�����v�Ƃ����p��̒�`�����̂��Ƃ��Ӗ����A���`�ł���
GAMP 4 �܂ł́A�\�t�g�E�F�A�̌��i���Ȃ킿�e�X�g�j�̂��Ƃ��o���f�[�V�����ƌĂB
�V�K�C�h���C���ɂ����Ă�������`�ł���B
�������Ȃ���AGAMP 5 �́AFDA ������PAT �Ƃ����T�O����т��̃C���v�������g�ł���uASTM E2500�v�ɐ��������A�o���f�[�V�����Ƃ����p����u�Ӑ}�����p�r�ւ̓K�����̒B���v�ƒ�`���Ă���B
����Ɍĉ����āAGAMP 4 �̃^�C�g���́A�uGAMP Guide for Validation of Automated Systems�v�ł��������AGAMP 5 �ł́uA Risk-Based Approach to Compliant GxP
Computerized Systems�v�ƂȂ�A�^�C�g������o���f�[�V�����Ƃ����p�ꂪ�������B�@
�����ł��܂��_�u���X�^���_�[�h�̌��O��������\��������B
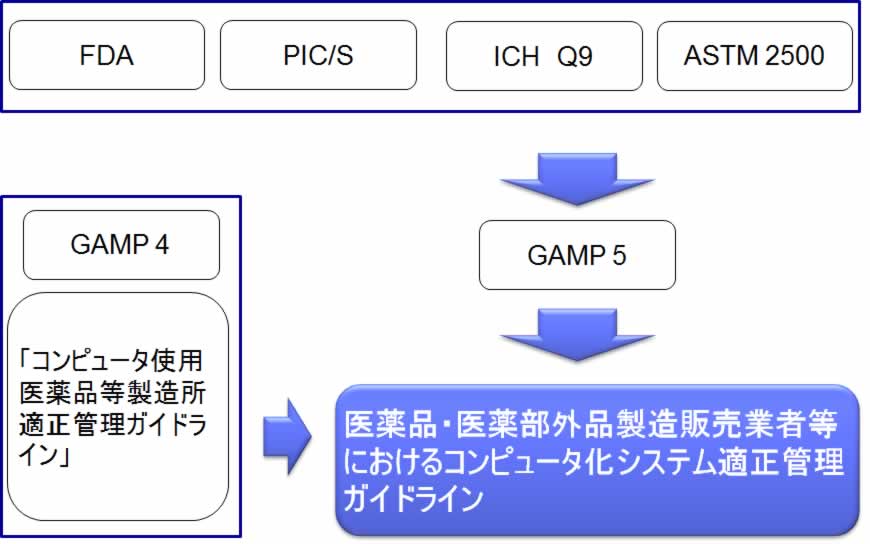
3.�@�K�C�h���C���̐V����r
�V�K�C�h���C���́A�ꌩ����Ƌ��K�C�h���C���Ǝ��Ă���悤�Ɍ�����B
�������Ȃ���A���̓��e�͑傫���قȂ�B�i�\1 �Q�Ɓj
�R���s���[�^�g�p���i���������K���Ǘ��K�C�h���C�� |
���i�E��O�i�����̔��Ǝғ��ɂ�����R���s���[�^���V�X�e���K���Ǘ��K�C�h���C���i�āj |
|---|---|
��1�@�ړI ��2�@�K�p�͈̔� |
1.�@���� 1.1�@�ړI |
��3�@�J���Ɩ� 1.�@�J�������i�K �i1�j�@�J���i�K�̐ӔC�̐��̊m�� �i2�j�@�J���}�j���A���̍쐬 �i3�j�@�J���v�揑�̍쐬 2.�@�V�X�e���v�i�K �i1�j�@�V�X�e���v���̍쐬 �i2�j�@�V�X�e���v���̊m�F 3.�@�v���O�����v�i�K �i1�j�@�v���O�����d�l���̍쐬 �i2�j�@�v���O�����d�l���y�уv���O�����̊m�F �i3�j�@�v���O�����e�X�g�̎��{ 4.�@�V�X�e���e�X�g�i�K �i1�j�@�V�X�e���e�X�g���{�v�揑�̍쐬 �i2�j�@�V�X�e���e�X�g�̎��{ 5.�@�ݒu�E�^�p�e�X�g�i�K �i1�j�@�ݒu�v�揑�̍쐬 �i2�j�@�n�[�h�E�F�A�̐ݒu �i3�j�@�^�p�e�X�g���{�v�揑�̍쐬 �i4�j�@�^�p�e�X�g�̎��{ |
4.�@�J���Ɩ� 4.1�@�J���v�揑�̍쐬 4.2�@�v���d�l���̍쐬 4.3�@�V�X�e���A�Z�X�����g 4.4�@�@�\�d�l���̍쐬 4.5�@�v�d�l���̍쐬 4.5.1�@�n�[�h�E�F�A�v�d�l 4.5.2�@�\�t�g�E�F�A�v�d�l 4.6�@�v���O�����̍쐬�y�уv���O�����e�X�g 4.6.1�@�v���O�����̍쐬 4.6.2�@�v���O�����e�X�g�̌v��y�ю��{ 4.7�@�V�X�e���e�X�g 4.7.1�@�V�X�e���e�X�g�v�揑�̍쐬 4.7.2�@�V�X�e���e�X�g�̎��{ 4.8�@������� |
5. ���؋Ɩ� 5.2�@�o���f�[�V�����S�̌v�揑�̍쐬 5.2.1�@�v���K�i���]���v�揑�̍쐬 5.2.2�@�v���K�i���]���̎��{ 5.2.3�@�v���K�i���]�����̍쐬 5.3�@���t���K�i���iIQ�j 5.4�@�^�]���K�i���]���iOQ�j 5.5�@���\�K�i���]���iPQ�j 5.6�@�K�i���]���̈ꕔ�ȗ��ƈ��p 5.7�@�o���f�[�V�����������̍쐬 |
|
��4�@�^�p�Ǘ��Ɩ� 1.�@���� �i1�j�@�^�p�Ǘ��i�K�̐ӔC�̐��̊m�� �i2�j�@�^�p�Ǘ��菇���̍쐬 �i3�j�@�V�X�e���̕ύX 2.�@�n�[�h�E�F�A�̑��� 3.�@�ێ�_�������̎��{ 4.�@���̔������̑Ή� 5.�@�Z�L�����e�B�Ǘ��̎��{ 6.�@���ȓ_���̎��{ |
6. �^�p�Ǘ��Ɩ� 6.1�@�^�p�Ǘ��ɂ�����ӔC�̐��̊m�� 6.2�@�^�p�Ǘ��̎菇�Ɋւ��镶���̍쐬 6.3�@�R���s���[�^�V�X�e���̑��� 6.4�@�ێ�_�������̎��{ 6.5�@�Z�L�����e�B�Ǘ��̎��{ 6.6�@�o�b�N�A�b�v�y�у��X�g�A 6.7�@�ύX�̊Ǘ� 6.8�@��E�̊Ǘ� 6.9�@����P�� 6.9.1�@����P���v��̍쐬 6.9.2�@����P���̎��{ |
7.���ȓ_�� 7.1�@���ȓ_���̎��{ 7.2�@���P�[�u�̎��{ |
|
8. �R���s���[�^�V�X�e���̔p�� 8.1�@�R���s���[�^�V�X�e���̔p���v�揑�̍쐬 8.2�@�R���s���[�^�V�X�e���̔p���L�^�̍쐬 |
|
��5�@�����y�ыL�^�̕ۑ��Ǘ� |
9. �����y�ыL�^�̊Ǘ� |
10. �p��W |
�@�@�@�@�@�@�@�@�@�@�@�@�@�@�@�@�\1�@�K�C�h���C���V����r
�ȉ��ɁA�V�K�C�h���C���Œlj��ƂȂ����A��v�ȗv�����L�ڂ���B
1)�u �R���s���[�^���V�X�e���Ǘ��K��v���̍쐬
2) �g�D�E�����ɉ������ӔC�ƌ����̖��m��
3) ���J��ER/ES �w�j�̗v���̏���
4) ��ړI�ȃo���f�[�V�����̎��{
5) �V�X�e���䒠�̍쐬
6) �v���d�l���̍쐬
7) �V�X�e���A�Z�X�����g�̎��{
�E�\�t�g�E�F�A�̃J�e�S������
�E���i�i���ɑ��郊�X�N�A�Z�X�����g
�E�����҃A�Z�X�����g
8) ���؋Ɩ��iDQ�AIQ�AOQ�APQ�j�̎��{
9) ���P�[�u�̎��{
10) �R���s���[�^�V�X�e���̔p��
11) �Ɩ��̌p�����̂��߂̗v���A��Q��̗v���A�f�[�^�̃o�b�N�A�b�v�A�A�N�Z�X�����A�A�N�Z�X�L�^��
3.1�@�u�R���s���[�^���V�X�e���Ǘ��K��v���̍쐬
�V�K�C�h���C���ł́A�u�R���s���[�^���V�X�e���Ǘ��K��v����CSV SOP ���쐬���邱�Ƃ����߂Ă���B
��ʂɁACSV SOP �́A�ŏ�ʂ�CSV Policy ���쐬���A�o�c�҂����F����B
Policy �ɂ́A�R���s���[�^���V�X�e���̊J���A���A�^�p�y�єp���Ɋւ����{���j��A�̐�����і����ƐӔC���L��
���Ȃ���Ȃ�Ȃ��B
�܂�Policy �̉��ʂɂ́A�菇���Ɗe��`�F�b�N���X�g���쐬���Ȃ���Ȃ�Ȃ��B
�菇���ɂ́A�쐬���ׂ������y�т��̊Ǘ����@��Ɩ������̊m�F�y�я��F�̎葱�����L�ڂ���B
�`�F�b�N���X�g�́A�V�X�e���A�Z�X�����g�⋟���Ҋč��̍ۂɕK�v�ƂȂ�̂ŏ������K�v�ł���B
3.2�@�g�D�E�����ɉ������ӔC�ƌ����̖��m��
CSV �����{����ɂ������Ă̑g�D����і����ƐӔC�m�ɂ��Ă����Ȃ���Ȃ�Ȃ��B
�������A�i���ۏɊւ���ӔC�̐��́AGQP �ȗ߂�GMP�ȗ߂ňقȂ邽�߁A�o���ɂ܂������đ̐����`����ꍇ��
�́A���ӂ��K�v�ł���B
3.3�@���J��ER/ES �w�j�̗v���̏���
�u���i���̏��F���͋����ɌW��\�����Ɋւ���d���I�L�^�E�d�q�������p�̂��߂̎w�j�v�i��������J��ER/ES�w�j�j�́A�����P�V�N�S���P���ɋǒ��ʒm�Ƃ��Ĕ��o���ꂽ�B
���o���炷�ł�5 �N�ȏオ�o�߂��Ă���B
ANNEX 11�i����Ńh���t�g�j �ɂ́A�d�q�L�^�E�d�q�����Ɋւ���v�������邽�߁A���������Ƃ邽�߂ɖ{�v���������ꂽ���̂Ɛ��@����B
�č��ł�1997 �N��21 CFR Part 11 ����������A����܂ő����̐����Ђ����̑Ή��ɋꗶ���Ă������Ƃ͎��m�̎�
���ł���B
���{�ł�Part11 �Ƃ��Ă��A���J��ER/ES �w�j�ɏ������邽�߂ɂ́A�����̓w�͂��K�v�ƂȂ�Ǝv����B
3.4�@��ړI�ȃo���f�[�V�����̎��{
PIC/S �̃K�C�_���X�ɂ́A��ړI�o���f�[�V�����̋L�ڂ��݂��邽�߁A���������Ƃ������̂Ɛ��@����B
��ړI�o���f�[�V�����́A���K�C�h���C�����ɏ]���ăo���f�[�V�������Ă��Ȃ��V�X�e���ɂ��āA��ړI�ɕi���⏞
���s���Ƃ��������̂ł���B
���݉ғ����ɃV�X�e����ݔ��Ɋւ��āA�e�X�i���ۏ��ł��Ă��邩���ēx�������Ă����Ȃ���Ȃ�Ȃ��B
���̋@��ɃV�X�e���̒I�����s���Ă����ׂ��ł���B
�����ɂ����āu��ړI�o���f�[�V�����v�ł͂Ȃ��A�u��ړI�ȃo���f�[�V�����v�ł��邱�Ƃɒ��ӂ��K�v�ł���B
�V�K�C�h���C���i�āj�ƂƂ��ɔ��\���ꂽ�u�l�����v�̖�31 �ɂ́A�u��ړI�ȃo���f�[�V���������{����ꍇ�A��̓I
�ɂ͂ǂ̂悤�Ɏ��{����悢�̂��v�Ƃ������₪����A�ȉ��̂悤�ɉ��Ă���B
�u��ړI�ȃo���f�[�V�����ɂ��ẮA���Y�V�X�e���̊J�����̎d�l���Ȃǂ̕����ނ�L�^�ނɑk���āA���̓K�i����
��������@��A���݂̎g�p�ړI�ɓK�������v���d�l�₻��ɏ����镶���Ƃ̓K�i�����m�F������@�����l�����邪�A
�K�i���̊m�F�ɂ������ẮA���݂̉^�p�ɂ�����L�^�ނ̏ƍ������I���r���[�̌��ʂ𗘗p���Ă��悢�B�v
�܂�A��ړI�ȃo���f�[�V�����ɂ�3 �̕��@���l������B
1) �ߋ��̎d�l��������B�ꍇ�ɂ���Ă͂������č쐬����B
2) ���݂̃V�X�e���̗��p���@�ɓK������悤�A���[�U�v���d�l����V�K�쐬����B
3) �ғ���̎��f�[�^���ƍ����A��肪�Ȃ����Ƃ��m�F����B
��L�̂����A3) ���{���̉�ړI�o���f�[�V�����ł���B
�V�K�o���f�[�V�����iProspective Validation�j�̏ꍇ�A�V�X�e���͂܂��ғ����Ă��Ȃ����߁A���f�[�^�͂Ȃ��A�e�X�g
�f�[�^��p�����������،^�̃e�X�g�����{���邱�ƂɂȂ�B
����ɑ��āA��ړI�o���f�[�V�����iRetorospective Validation�j�ł́A���łɉғ����Ă��邱�Ƃ���A�e�X�g�f�[
�^�ł͂Ȃ��A��茻���I�Ȏ��f�[�^�������邱�Ƃɂ��A���Y�V�X�e���̐M�������m�F���邱�Ƃ��ł���B����������
��ʂɉ�ړI�o���f�[�V�����ł́A�e�X�g�����{���Ȃ��B
�����A�ғ����Ă���V�X�e�����ēx�e�X�g���邱�Ƃ���ړI�o���f�[�V�����ƌĂ�ł���̂����ɂ��邪�A������
�͍ăo���f�[�V�����iRe-Validation�j�ƌĂԁB
3.5�@�V�X�e���䒠�̍쐬
�V�X�e���䒠�́A��ʂɉp��ł̓V�X�e���C���x���g���ƌĂ��B
����͎{�ݓ���H����ɐݒu����Ă���R���s���[�^���V�X�e�����ꗗ�\�ɂ܂Ƃ߂����̂ł���B
���Ă̓��ǂ́A�ȑO���炱�̃V�X�e���C���x���g���̍쐬���`���t���Ă����B
�R���s���[�^���V�X�e���̍��@����ۂɂ́A���̃V�X�e���C���x���g������A���Y�{�ݓ���H����ɂǂ̂悤
�ȃR���s���[�^���V�X�e�������݂���̂���ł��Ȃ���Ȃ�Ȃ�����ł���B
�V�X�e���C���x���g���ɂ́AH/W �̖��́AS/W �̖��̂ƃo�[�W�����A�쐬����SOP ��o���f�[�V�����̋L�^���L��
���Ă����Ȃ���Ȃ�Ȃ��B
�܂��V�X�e�����X�N�A�Z�X�����g�ɂ���Ĕ��肳�ꂽ�A���X�N�ɂ��Ă��L�ڂ��Ă����K�v������B
����ɃV�X�e���C���x���g�����쐬����ۂ́A�Œ莑�Y�䒠�Ȃǂ��Q�l�ɂ���Ɨǂ����낤�B
�����Œ��ӂ��K�v�Ȃ��Ƃ́A�l�Ƀp�\�R���ɃC���X�g�[������Ă���Excel ���̃A�v���P�[�V�����ł���B
Excel �͈�ʂ̗ʔ̓X�ł��w���ł�������ȃA�v���P�[�V�����ł��邽�߁A�����Ƃ������ł��邪�A������A�i����
���̋L�^�A�o�ה���̋L�^�A�����L�^���̃��X�N�̍����L�^�ޓ���ێ����Ă�����A�Ǘ����Ă���ꍇ�͗v���ӂł���B
�V�X�e���̉��i�ɂ�����炸�A�����d�q�L�^�̏d�v����X�N�ɉ������Ή����d�v�Ƃ�����B
�V�X�e���C���x���g�����쐬����ۂɂ́A���Y����ɂ�����Excel �̗��p�ɂ��Ă��O��I�ɒ������Ă������Ƃ��̐S
�ł���B
3.6�@�v���d�l���̍쐬
�v���d�l���́A��ʓI�ɂ̓��[�U�v���d�l���ƌĂ��B
���K�C�h���C���ł́A���[�U�v���d�l���̍쐬���v������Ă��Ȃ������B
�������Ȃ���A���Ă̋K���v����GAMP ���ł́A���[�U�v���d�l���̍쐬�͕K�{�ł���B
�Ȃ��Ȃ�A�o���f�[�V�����Ƃ́A���[�U�̗v��������悤�R���s���[�^���V�X�e�����J���A�����A�^�p���邱��
�ł��邩��ł���B
�i���ۏ̊�{�����́A���炩���ߒ�߂��d�l��i�������݂��A�ŏI�I�ɂ������������Ƃ��ؖ�����؋�������
���Ȃ���Ȃ�Ȃ��̂ł���B
3.7�@�V�X�e���A�Z�X�����g�̎��{
�V�X�e���A�Z�X�����g�ł́A�ȉ���3 �����{����悤�ɋ��߂Ă���B
1) �\�t�g�E�F�A�̃J�e�S������
���炩���߃\�t�g�E�F�A�̃J�e�S�����ނ����{���Ȃ���Ȃ�Ȃ��B
�\�t�g�E�F�A�̃J�e�S�����ނ́AGAMP �ł悭�m���Ă���B
���Ă̋K���v���ɂ́A���̃\�t�g�E�F�A�̃J�e�S�����ނ͂Ȃ��A�{�K�C�h���C���ɓ����I�ł���B
�������Ȃ���A�\�t�g�E�F�A�̃J�e�S�����ނ́A�T�v���C���Ƌ��c���A�v���ł܂�Ȃ��ƌ���ł��Ȃ��Ƃ�����肪��
��A�J���Ɩ��̏����i�K�ł͌���͍���ł���B
�\�t�g�E�F�A�̃J�e�S�����ނ̏ڍׂɂ��ẮAGAMP 5 ���w�K���Ă����K�v������B
2) ���i�i���ɑ��郊�X�N�A�Z�X�����g
���Y�R���s���[�^���V�X�e�������Y�����ܓ��̐��i�̃��X�N�肵�Ȃ���Ȃ�Ȃ��B
���Ȃ킿�A���Y���i�̕i���Ɉُ킪�������ۂ̊��҂ɗ^���錒�N��Q�̃��X�N�����炩���ߒ������Ă����̂ł���B
���X�N�A�Z�X�����g�Ɋւ���ڍׂ́AICH Q9 ���w�K���Ă����Ȃ���Ȃ�Ȃ��B
3) �����҃A�Z�X�����g
�R���s���[�^���V�X�e���̕i���́A���Y�T�v���C���ɂ��Ƃ��낪�傫���B
���������āA�T�v���C���̒�������ёI�肪�d�v�ł���Ƃ�����B
�T�v���C���̑I��ɂ������ẮA�T�v���C���I�[�f�B�b�g�����{���Ȃ���Ȃ�Ȃ��B
�T�v���C���I�[�f�B�b�g�́AAd-hoc �Ɏ��{���ׂ��ł͂Ȃ��A�`�F�b�N���X�g��p���āA�\���Ɍv�悵����Ŏ��{����K�v������B
3.8�@���؋Ɩ��iDQ�AIQ�AOQ�APQ�j�̎��{
���K�C�h���C���ł́A���؋Ɩ��Ɋւ���L�q���Ȃ������B
�V�K�C�h���C���ł́AGAMP 4 �ɏ]���A�܂����݂̂Ƃ��뉢�Ă𒆐S�ɑ������{����Ă���DQ�AIQ�AOQ�APQ �ƌĂ�
��錟�ؕ��@��p���Ă���B
�������Ȃ���A�O�q�����Ƃ���A�ŐV�̉��Ă̋K���v����GAMP 5 �ł́ADQ�AIQ�AOQ�APQ �Ƃ����p����g�p���Ȃ��Ȃ������߁A������{��Ƃɂ����ẮA�_�u���X�^���_�[�h�̖�肪���O�����B
3.9�@���P�[�u�̎��{
���P�[�u�ɂ��Ă��A�V�K�C�h���C���ɐV�����lj����ꂽ�v�������ł���B
FDA �́A2004 �N����ACAPA�iCorrective Actions;
Preventive Actions�F�\�h�[�u�E�����[�u�j�Ɋւ��鍸�@��
���������o�܂�����B
3.10�@�R���s���[�^�V�X�e���̔p��
�R���s���[�^�V�X�e���̔p���ɂ��Ă��A�V�K�C�h���C���Œlj��ƂȂ����B
�R���s���[�^�V�X�e����p������ۂɂ́AH/W ��S/W �͔p�����Ă��\��Ȃ����A���Y�V�X�e���̑���菇���A�^�p�}
�j���A�����̎菇���ނ�ACSV �̋L�^���͎̂ċ����Ă͂Ȃ�Ȃ��B
�܂����Y�R���s���[�^�V�X�e�����ێ����Ă���A�d�q�L�^��d�q�����́A���m�����S�Ɉ��S�ȏꏊ�ɃA�[�J�C������
�����Ȃ���Ȃ�Ȃ��B
�����Ŋ��S�Ƃ́A�č��ؐՓ��̃��^�f�[�^���܂߂āA�ڍs���Ă����Ƃ����Ӗ��ł���B
���̂����ŁA�d�q�L�^�E�d�q�����́A���@�����{���ꂽ�ۂɁA���@���ɒł���悤�A�����ƈ���̋@�\��ۗL����
���Ȃ���Ȃ�Ȃ��B
3.11�@�Ɩ��̌p�����̂��߂̗v���A��Q��̗v���A�f�[�^�̃o�b�N�A�b�v�A�A�N�Z�X�����A�A�N�Z�X�L�^��
�V�K�C�h���C���i�āj�ƂƂ��ɔ��\���ꂽ�u�l�����v��Q33 �ɂ́A�u�R���s���[�^�V�X�e����������v���v�ɂ���
�̋L�ڂ�����B
�u�{�K�C�h���C���̑ΏۂƂȂ�R���s���[�^�V�X�e���ɂ́A�ǂ̂悤�ȗv���������K�v������̂��B�v�Ƃ����₢
�ɑ��A�ȉ��̂悤�ȗv�������Ă���B
• ER/ES �w�j�̗v��
• �^�����A���ǐ��A�ۑ����m�ۂ̂��߂̗v��
• �Ɩ��̌p�����̂��߂̗v��
• �Z�L�����e�B�m�ۂ̂��߂̗v��
• �o�b�N�A�b�v�A�A�N�Z�X�����A�A�N�Z�X�L�^���Ɋւ���v��
• �o�b�N�A�b�v���f�B�A�ɋ��߂���v��
• ���f�B�A�̕ۑ����@�̗v��
�����̗v����ANNEX 11 �Ƃ̐������������������̂ł���Ɛ��@����邪�A�ł͂Ȃ��K�C�h���C���{�̂ɋL�ڂ���
���Ȃ��̂ł��낤���B
�u�l�����v��Q34 �ł́A�Ɩ��p�����Ƃ͂ɂ��Ĉȉ��̂悤�ɉ��Ă���B
�Ɩ��p�����Ƃ́A�̏��V�X�e���g���u���ɂ��A�Ɩ��̒��f������邽�߁A����̂��߂̑[�u���u������A�g���u��
���������Ă��A���Y�Ɩ����p���\�ȑ[�u��p�ӂ���Ȃǂ̑Ή����s�����Ƃł���B
�Ɩ��̌p�����m�ۂ̂��߂̗v���Ƃ�
• �V�Ђ�l�דI�W�Q���l�������ݒu�����̐ݒ�
• �f�[�^�̃o�b�N�A�b�v�i�o�b�N�A�b�v�̕��@��ۑ����@���j
• ����̑�@�����炩���ߗp��
• �\�߃}�j���A���ɂ���֎菇���K�肵�Ă���
• �v���ȃV�X�e���̕����̂��߂̑[�u�̎菇
• ����I�Ƀf�[�^�̃o�b�N�A�b�v��ۑ�
�Ȃǂł���B
�Ɩ��p�����̕K�����́A���X�N�A�Z�X�����g�̌��ʓ����l�����ē��Y���X�N�ɉ����Č��肷�邱�ƂɂȂ�B
4.�@�V�K�C�h���C���Ή��̗��ӓ_
�V�K�C�h���C���ɑΉ����邽�߂ɗ��ӂ��Ȃ���Ȃ�Ȃ��Ǝv���鎖�����L�ڂ���B
�P�D�_�u���X�^���_�[�h�ւ̑Ή�
��q�����ʂ�A�O���[�o����CSV�Ɋւ���p��ƐV�K�C�h���C���̗p��ł́A���̎g�������`�ɈႢ������B���������ăO���[�o����ƂɂƂ��ẮA�_�u���X�^���_�[�h�ɑΉ����Ȃ���Ȃ�Ȃ����ƂɂȂ�B
�Q�D�{�K�C�h���C���ɑ�����@��p���Ă��Ƃ��Ă��邪�A���{�̍��@�ɂ����ẮA�{�K�C�h���C�������ƂɎ��{�����̂ŁA���ӂ��K�v�ł���B
�R�D�\�t�g�E�F�A�̃J�e�S�����ނɂ��ẮAGAMP 5���Q�l�ɂ��Ă��邽�߁AGAMP
5���悭�������Ă����Ȃ���Ȃ�Ȃ��B�܂��\�t�g�E�F�A�̃J�e�S�����ނ̕��@�́A�e�ЂŌ��肵�A�R���s���[�^���V�X�e���Ǘ��K�蓙�ɋL�ڂ��Ă����Ȃ���Ȃ�Ȃ��B
�S�D��`�ɋL�ڂ���Ă��Ȃ��p��
GAMP�A�\�t�g�E�F�A�J�e�S���A���X�N�A�V�X�e���䒠���̗p��͒�`����Ă��Ȃ��B
�T�D���X�N�A�Z�X�����g�ɂ��ẮAICH-Q9���悭�������đΉ�����K�v������B
5.�@�V�K�C�h���C������
�P�D����
�P�D����
|
�{�K�C�h���C���́A���K�C�h���C���ł���u�R���s���[�^�g�p���i���������K���Ǘ��K�C�h���C���v��u����������̂ł���B
�P�D����
|
�u������@�v�Ƃ́AGAMP 5�����w���B
�������Ȃ���A���{�̍��@�ɂ����ẮA�{�K�C�h���C�������ƂɎ��{�����̂ŁA���ӂ��K�v�ł���B
1.2�@�R���s���[�^���V�X�e���̎戵�����̃K�C�h���C���́A�f�p�o�ȗߋy�тf�l�o�ȗ߂Ɋ֘A����V�X�e�����тɑ��݂ɘA�g�����R���s���[�^���V�X�e����ΏۂƂ��Ď�舵�����ƂƂ��Ă��邽�߁A�f�p�o�ȗ߂�f�l�o�ȗ߂ɂ�����g�D�E�����ɉ������\����p���Ă��Ȃ����A�o���f�[�V������ύX�E��E�̊Ǘ��ȂǁA�f�l�o�ȗ߂ɂ����Ă͕i�����哙�̏��F���K�v�ł���A�f�p�o�ȗ߂ɂ����Ă͕i���ۏؕ���ɂ��Ǘ��̐��̒��Ői�߂Ȃ���Ȃ�Ȃ��B�]���āA�����̔��Ǝғ��ɂ����đg�D�̌`�Ԃ�Y������V�X�e���͈̔͂��l�����Ċe�X�̑g�D�E�����ɉ������ӔC�ƌ������R.�ɋK�肷��u�R���s���[�^���V�X�e���̊J���A���؋y�щ^�p�̎菇���Ɋւ��镶���v�̒��ɖ��m�ɂ��邱�Ƃ��K�v�ł���B |
�g�D�E�����ɉ������ӔC�ƌ����m�ɂ���K�v������B
1.2�@�R���s���[�^���V�X�e���̎戵���i�����j�܂��A���̃K�C�h���C���̑ΏۂƂȂ�R���s���[�^���V�X�e���́u���i���̏��F���͋����ɌW��\�����Ɋւ���d���I�L�^�E�d�q�������p�̂��߂̎w�j�v(�����P�V�N�S���P����H����0401022��)�y�сu�@�y�э̌��y�ы���������Ǝ���@�̈ꕔ����������@���̎{�s�ɔ������i�C��Ë@�퓙�̐����Ǘ��y�ѕi���Ǘ��i�f�l�o�^�p�l�r�j�ɌW��ȗߋy�э����̐���y�щ��p�ɂ��āv (�����P�V�N3��30����H�Ė�����0330001��)��R�͑�R �R�T�D�u���̑��i�d���I�L�^���ɂ��āj�v�̗v���������K�v������B |
���J��ER/ES�w�j�ɏ������邱�ƁB
1.2�@�R���s���[�^���V�X�e���̎戵���i�����j�Ȃ��A���̃K�C�h���C���̎{�s���ȑO�ɊJ�����͉^�p���J�n����Ă���V�X�e���ł����āA�u�R���s���[�^�g�p���i���������K���Ǘ��K�C�h���C���v�Ɏ����ꂽ���@���͂���ɑ���K�ȕ��@�ŊJ���A���؋y�щ^�p�����s���Ă��Ȃ��V�X�e���ɂ��ẮA��ړI�ȃo���f�[�V�������ɂ��A���Y�V�X�e���̓K�i�����m�F����K�v������B |
��ړI�ȃo���f�[�V�����F���K�C�h���C���ɏ]���ĊJ���E�Ǘ�����Ă���̂��O���A�]�p���ꂽ�V�X�e�����̑Ή����K�v�B
1.3�@�J�e�S���������̃K�C�h���C���̓K�p����R���s���[�^���V�X�e���ɂ��ẮA�J���A���؋y�щ^�p�̊e�i�K�ɂ����Ď��{������e������(�u4.3�V�X�e���A�Z�X�����g�v���Q��)���邽�߂ɁA�V�X�e�����\������\�t�g�E�F�A�̎�ނɉ����āA���炩���߃\�t�g�E�F�A�J�e�S�������肷����̂Ƃ���B �J�e�S�����ނ̊�y�уJ�e�S�����̈�ʓI�Ή��̗��ʎ��Q�u�J�e�S�����ޕ\�v�Ɏ������B |
�{�K�C�h���C���ł́AGAMP�Ɠ��l�A�\�t�g�E�F�A���J�e�S���������Ă���B
�J�e�S���͂��Ȃ킿�\�t�g�E�F�A�̃��X�N�̕��ނł���B�J�e�S���̐������傫���Ȃ�قǁA���[�U�ɂ��ύX�̓x�������傫���A�\�t�g�E�F�A�Ɍ��ׂ��܂܂��\�������傷��B
���X�N�A�Z�X�����g�̌��ʂƂ��Ă��ȕւȎ戵���s�Ȃ��Ă������x���Ȃ��Ƃ��鍇���I�ȍ������������Ƃ��ł���悤�ɂ��Ȃ���Ȃ�Ȃ��B
�Q�D�K�p�͈̔�
2.�@�K�p�͈̔����̃K�C�h���C���́A�f�p�o�ȗߋy�тf�l�o�ȗ߂��K�p�����Ɩ��ɁA�R���s���[�^���V�X�e�����g�p���鐻���̔��Ǝғ��ɓK�p����B ���̃K�C�h���C���̑ΏۂƂȂ�R���s���[�^���V�X�e���̗�Ƃ��āA�ȉ����l������B (1) ���i�A��O�i�̎s��ւ̏o�ׂ̉ۂ̌���ɌW��V�X�e���y�юs��ւ̏o�ׂɌW��L�^���쐬�A�ۑ��Ǘ����邽�߂̃V�X�e�� (2) �����w�}���A�����Ɋւ���L�^�����쐬�y�ѕۑ��Ǘ����邽�߂̃V�X�e�� (3) �����H���𐧌䖔�͊Ǘ����邽�߂̃V�X�e���y�т��̊Ǘ��f�[�^��ۑ��Ǘ����邽�߂̃V�X�e�� (4) ���ޗ��y�ѐ��i�i�����̒��ԍH���ő�������̂��܂ށB�ȉ������B�j�̕ۊǁA�o�[���̐��Y���Ǘ�����V�X�e�� (5) �i�������̂��߂̋@��𐧌�E�Ǘ�����V�X�e�����тɕi���������ʋy�ъǗ��f�[�^��ۑ��Ǘ����邽�߂̃V�X�e�� (6) �A�����p�������ݔ��ȂǁA���i�̕i���ɏd��ȉe�����y�ڂ��\���̂��鐻���x���ݔ��E�{�݂𐧌䖔�͊Ǘ����邽�߂̃V�X�e���y�т��̊Ǘ��f�[�^��ۑ��Ǘ����邽�߂̃V�X�e�� (7) �����i�菇���ށA�i���W�����A���i�W�������j���쐬�A���F�A�ۑ��Ǘ����邽�߂̃V�X�e�� |
�R�D�R���s���[�^���V�X�e���̊J���A���؋y�щ^�p�̎菇���Ɋւ��镶���̍쐬
3.�@�R���s���[�^���V�X�e���̊J���A���؋y�щ^�p�̎菇���Ɋւ��镶���̍쐬�����̔��Ǝғ��̓R���s���[�^���V�X�e���̊J���A���؋y�щ^�p�ɂ������ẮA���炩���߁A���̎菇���Ɋւ��镶���i�ȉ��u�R���s���[�^���V�X�e���Ǘ��K��v�Ƃ����B�j���߂���̂Ƃ���B �R���s���[�^���V�X�e���Ǘ��K��́A�����Ƃ��Ď��̎������L�ڂ�����̂Ƃ��� (1) �R���s���[�^���V�X�e���̊J���A���؋y�щ^�p�Ɋւ����{���j �|�ړI �|�K�p�͈� �|�V�X�e���䒠�̍쐬 �|��{�I�ȍl���� �E�\�t�g�E�F�A�̃J�e�S������ �E���i�i���ɑ��郊�X�N�A�Z�X�����g �E�����҃A�Z�X�����g �E�J���A���؋y�щ^�p�i�K�Ŏ��{���ׂ����ړ� (2) �J���Ɩ��A���؋Ɩ��y�щ^�p�Ǘ��Ɩ��ɂ�����ӔC�̐��Ɩ��� (3) �J���Ɩ��A���؋Ɩ��y�щ^�p�Ǘ��Ɩ��ō쐬���ׂ������y�т��̊Ǘ����@ (4) �J���Ɩ��A���؋Ɩ��y�щ^�p�Ǘ��Ɩ��̋Ɩ������̊m�F�y�я��F�̎葱�� |
�R���s���[�^���V�X�e���Ǘ��K��͊�ƑS�̂œ��ꂵ�������Ƃ��Đ��肷��ꍇ�����邵�A�X�̕���Ōʂɐ��肷��ꍇ������B
���ꂵ�������𐧒肷��ꍇ�ł����Ă��A�f�p�o��f�l�o�̊�ɉ����ĕ����̏��F���s�����Ƃ��K�v�ł���B
�X�ɐ��肷��ꍇ�́A�K�v�ɉ����ĕ���Ԃł̊�{�I�戵���̐������̊m�ۂƁA����Ԃł̖������S�ƘA�g�̕��@�ɂ��ċL�ڂ��Ă������Ƃ��K�v�ł���B
�S�D�J���Ɩ�
4.�@�J���Ɩ�
4.1�@�J���v��Ɋւ��镶���̍쐬�����̔��Ǝғ��́A�J���v��Ɋւ��鎖�����L�ڂ��������i�ȉ��u�J���v�揑�v�Ƃ����B�j���쐬����B�J���v�揑�ɂ́A�����Ƃ��Ď��̎������L�ڂ�����̂Ƃ���B (1) �J���̖ړI (2) �J������ (3) �J���̐� �|�g�D �|�ӔC�� �E�J���ӔC�� �E���ؐӔC�� (4) �J���X�P�W���[�� |
4.2�@�v���d�l�Ɋւ��镶���̍쐬�J���ӔC�҂̓R���s���[�^���V�X�e���ɋ��߂��Ă��鎖�����L�ڂ��������i�ȉ��u�v���d�l���v�Ƃ����B�j���쐬����B�v���d�l���ɂ͌����Ƃ��Ď��̎������L�ڂ�����̂Ƃ��� (1) �K�p����@�K���� (2) �n�[�h�E�F�A�̊T�v (3) �v���@�\ �|�V�X�e���̋@�\�̊T�v �|�^�p�v���̊T�v �|���\�v���̊T�v �|��Q��@�\�̊T�v �|�@���ی�@�\�̊T�v�i�Z�L�����e�B�j (4) �f�[�^ �|���o�͏��̍��ڂ̈ꗗ �|�ۑ����@ (5) �C���^�[�t�F�[�X�i�֘A�ݔ��y�ё��V�X�e�����j (6) �� �|�ݒu���� �|�V�X�e���̔z�u (7) �d���A�ڒn���̐��t���� |
4.3�@�V�X�e���A�Z�X�����g�J���ӔC�҂͊J���A���؋y�щ^�p�̊e�i�K�ɂĎ��{���ׂ����ړ������肵�A���ꂼ��̓��e���߂邽�߂ɁA�R���s���[�^���V�X�e���Ǘ��K��Ɋ�Â��ȉ��̎��������{����B (1) �\�t�g�E�F�A�J�e�S������ (2) ���i�i���ɑ��郊�X�N�A�Z�X�����g (3) �����҃A�Z�X�����g |
�{�K�C�h���C���ł́AGAMP�Ɠ��l�A�\�t�g�E�F�A���J�e�S���������Ă���B
�J�e�S���͂��Ȃ킿�\�t�g�E�F�A�̃��X�N�̕��ނł���B�J�e�S���̐������傫���Ȃ�قǁA���[�U�ɂ��ύX�̓x�������傫���A�\�t�g�E�F�A�Ɍ��ׂ��܂܂��\�������傷��B
���X�N�A�Z�X�����g�̌��ʂƂ��Ă��ȕւȎ戵���s�Ȃ��Ă������x���Ȃ��Ƃ��鍇���I�ȍ������������Ƃ��ł���悤�ɂ��Ȃ���Ȃ�Ȃ��B
4.4�@�@�\�d�l�Ɋւ��镶���̍쐬�J���ӔC�҂́A�����҂ɗv���d�l���ɋL�ڂ��ꂽ�v���ɑΉ�������̓I�ȃR���s���[�^���V�X�e���̋@�\�Ɛ��\���L�q�����@�\�d�l���L�ڂ��������i�ȉ��u�@�\�d�l���v�Ƃ����B�j���쐬�����A���F������̂Ƃ���B |
4.5�@�v�d�l�Ɋւ��镶���̍쐬�J���ӔC�҂́A�����҂ɋ@�\�d�l�Ɋ�Â��ăR���s���[�^���V�X�e���̏ڍ@�\���L�q�����v�d�l�Ɋւ��镶���i�ȉ��u�v�d�l���v�Ƃ����B�j���쐬�����A���F������̂Ƃ���B �v�d�l���ɂ́A�����Ƃ��Ď��̎������L�ڂ�����̂Ƃ���B 4.5.1 �n�[�h�E�F�A�v�d�l(1) �n�[�h�E�F�A�\�� (2) �n�[�h�E�F�A���X�g�y�юd�l (3) �C���^�[�t�F�[�X (4) ���o�͐M���̏ڍ� (5) �� �|�ݒu�̏ڍ��� �|�V�X�e���@��̔z�u (6) �d���A�ڒn���̐��t���� 4.5.2 �\�t�g�E�F�A�v�d�l(1) ���o�͏��̏ڍ� (2) �t�@�C���y�уf�[�^�\�� (3) �f�[�^�����̏ڍ� (4) �@�\�E���W���[���̍\�� (5) �C���^�[�t�F�[�X�̏ڍ� (6) �I�������p�b�P�[�W�\�t�g�E�F�A |
4.6�@�v���O�����̍쐬�y�уv���O�����e�X�g�J���ӔC�҂́A�K�v�ɉ����āA�����҂Ƀv���O�����쐬�y�уv���O�����e�X�g�����{��������̂Ƃ��顃v���O�����쐬�y�уv���O�����e�X�g�ɂ́A�ȉ��̓��e���܂܂����̂Ƃ���B 4.6.1 �v���O�����̍쐬(1) �����҂́A�v���O�����̎d�l�Ɋւ��镶���i�ȉ��u�v���O�����d�l���v�Ƃ����B�j��v�d�l���ɏ]���č쐬������̂Ƃ��� (2) �����҂̓v���O�������v���O�����d�l���ǂ���ɍ쐬������̂Ƃ��� 4.6.2 �v���O�����e�X�g�̌v��y�ю��{(1) �����҂́A�v���O�����e�X�g���@�A�v���O�����e�X�g���ʂ̔�����@�y�є������L�ڂ����v���O�����e�X�g�̌v��Ɋւ��镶���i�ȉ��u�v���O�����e�X�g�v�揑�v�Ƃ����B�j���쐬������̂Ƃ��� (2) �����҂́A�v���O�����e�X�g�v�揑�Ɋ�Â��A�v���O�����e�X�g�����{���A���̌��ʂ��L�^������̂Ƃ���B (3) �����҂́A�v���O�����e�X�g�̌��ʂ̓K�ۂ肷����̂Ƃ���B |
4.7�@�V�X�e���e�X�g�J���ӔC�҂́A�K�v�ɉ����ċ����҂ɃV�X�e���e�X�g�����{��������̂Ƃ��顃V�X�e���e�X�g�ɂ͈ȉ��̓��e���܂܂����̂Ƃ���B 4.7.1 �V�X�e���e�X�g�̌v��Ɋւ��镶���̍쐬�����҂̓V�X�e���e�X�g�ɂ������ẮA�V�X�e���e�X�g�̌v��Ɋւ��镶���i�ȉ��u�V�X�e���e�X�g�v�揑�v�Ƃ����B�j���쐬������̂Ƃ���B�V�X�e���e�X�g�v�揑�ɂ́A�����Ƃ��Ď��̎������L�ڂ�����̂Ƃ��� (1) �V�X�e���e�X�g�̎��{���i�e�X�g���̃n�[�h�E�F�A�̐ݒu�y�у\�t�g�E�F�A�\������������j (2) �V�X�e���e�X�g�̍��ڋy�юg�p����e�X�g�f�[�^ (3) �V�X�e���e�X�g�̕��@�y�ь��ʂ̊m�F���@ (4) �V�X�e���e�X�g�̔��� (5) �V�X�e���e�X�g�̃X�P�W���[�� (6) �V�X�e���e�X�g�����{����ꍇ�̎��{�̐� 4.7.2 �V�X�e���e�X�g�̎��{(1) �����҂́A�V�X�e���e�X�g�v�揑�Ɋ�Â��ăV�X�e���e�X�g�����{���A���̌��ʁi�V�X�e���e�X�g�̎��{���ɔ��������g���u���̓��e�y�т��̑[�u���e���܂ޡ�j���L�^������̂Ƃ��� (2) �����҂́A�V�X�e���e�X�g�̌��ʂ̓K�ۂ肷��B���̏ꍇ�ɂ����āA�V�X�e���e�X�g�̌��ʂ̓K�ۂ̔��莖���́A�����Ƃ��Ď��̂Ƃ���Ƃ��� �|�@�\�i�@�\�d�l���y�ѐv�d�l���ɋK�肳�ꂽ�Ƃ���ɋ@�\���邩���j �|���\�i�@�\�d�l���y�ѐv�d�l���Ŋ��҂��ꂽ�����������m�ۂ��Ă��邩���j |
4.8�@��������J���ӔC�҂́A�V�X�e���̋@�\�y�ѐ��\�̑S�Ă��邢�͈ꕔ���v���d�l�����Ă��邱�Ƃ��m�F���邽�߂ɋ����҂Ɏ�����������{������B��������ɂ́A�����҂̍H��o�בO�ɋ@�\�y�ѐ��\���m�F����e�X�g�i�H��o�����A�ȉ��u�e�`�s�v�Ƃ����B�j���тɂ����V�X�e���ݒu�ꏊ���ɂ�������ꎞ�ɋ@�\�y�ѐ��\���m�F����e�X�g�i���n��������A�ȉ��u�r�`�s�v�Ƃ����B�j������A�K�X�I�������{������B��������̌��ʂ͊J���ӔC�҂����F����B |
�T�D���؋Ɩ�
5.�@���؋Ɩ�
5.1�@�o���f�[�V�����̑S�̌v��Ɋւ��镶���̍쐬���ؒS���҂́A�R���s���[�^���V�X�e���Ǘ��K��Ɋ�Â��A�V�X�e���̌����s���ꍇ�ɂ́A���{����o���f�[�V�����̑S�̌v��Ɋւ��镶���i�ȉ��u�o���f�[�V�����v�揑�v�Ƃ����B�j���쐬������̂Ƃ���B�Ȃ��A�o���f�[�V�����v�揑�́u4.3�V�X�e���A�Z�X�����g�v�ɂ����{�����]�����ʓ��Ɋ�Â��쐬����B�o���f�[�V�����v�揑�͌��ؐӔC�҂̏��F����̂Ƃ���B�Ȃ��A���؋Ɩ��͊J���Ɩ��ƕ��s���čs���邱�Ƃ����邽�߁A�o���f�[�V�����v�揑�͊J���i�K�̓K�Ȏ����ɍ쐬����B �܂��A�u6.7�ύX�̊Ǘ��v�ɂ����ăo���f�[�V�������K�v�ƂȂ����ꍇ�́A�ύX�̏ɂ��킹�ēK�X�o���f�[�V�����v�揑���쐬���邱�ƁB�o���f�[�V�����v�揑�ɂ́A�����Ƃ��Ď��̎������L�ڂ�����̂Ƃ���B�܂��A�K�v�ȏꍇ�ɂ͏ڍׂȃ��X�N�A�Z�X�����g�A�����Ҋč����̌v��ɂ��Ă��L�ڂ��邱�ƁB |
5.�@���؋Ɩ�
5.1�@�o���f�[�V�����̑S�̌v��Ɋւ��镶���̍쐬�i�����j(1) �ړI (2) �V�X�e���T�v (3) �ӔC�̐��Ɩ��� �|�g�D �|���ؐӔC�� (4) �K�p����@�K���� (5) �o���f�[�V�������j �|�o���f�[�V�����͈̔͋y�уo���f�[�V�����Ƃ��Ď��{���ׂ����ړ� (6) �X�P�W���[�� (7) �o���f�[�V�������{���̕ύX�E��E�̊Ǘ��Ɋւ���菇 |
5.2�@�v���K�i���]���iDQ�j
5.2.1 �v���K�i���]���̌v��Ɋւ��镶���̍쐬���ؒS���҂́A�v���K�i���]���̌v��Ɋւ��镶���i�ȉ��u�v���K�i���]���v�揑�v�Ƃ����B�j���쐬���A���ؐӔC�҂̏��F����̂Ƃ���B�v���K�i���]���v�揑�ɂ́A�����Ƃ��Ď��̎������L�ڂ�����̂Ƃ��� (1) �v���K�i���]���ɌW�镶���� (2) ��̓I�Ȋm�F�̕��@ (3) �v���K�i���]���ɂ����锻�� (4) �X�P�W���[�� (5) �ӔC�ҋy�ђS���҂̎��� 5.2.2 �v���K�i���]���̎��{(1) ���ؒS���҂́A�v���K�i���]���v�揑�Ɋ�Â��ĕ]�������{���A���̌��ʂ��L�^������̂Ƃ��� (2) ���ؐӔC�҂́A�v���K�i���]���̌��ʂ̓K�ۂ肷����̂Ƃ���B 5.2.3 �v���K�i���]���̕Ɋւ��镶���̍쐬 ���ؒS���҂́A�v���K�i���]���̕Ɋւ��镶���i�ȉ��u�v���K�i���]�����v�Ƃ����B�j���쐬���A���ؐӔC�҂̏��F����̂Ƃ���B�v���K�i���]�����ɂ́A�����Ƃ��Ď��̎������L�ڂ�����̂Ƃ��� (1) �v���K�i���]���ɌW�镶���� (2) �]�����ʂƐ����[�u (3) �ӔC�ҋy�ђS���҂̎��� |
5.3�@���t���K�i���]���iIQ�j
5.3.1 ���t���K�i���]���̌v��Ɋւ��镶���̍쐬���ؒS���҂́A�n�[�h�E�F�A�y�у\�t�g�E�F�A�̐��t���K�i���]���̌v��Ɋւ��镶���i�ȉ��u���t���K�i���]���v�揑�v�Ƃ����B�j���쐬���A���ؐӔC�҂̏��F����̂Ƃ���B���t���K�i���]���v�揑�ɂ͌����Ƃ��Ď��̎������L�ڂ�����̂Ƃ��� (1) ���t���K�i���]���ɌW�镶���� (2) �n�[�h�E�F�A�\���y�ѐݒu�ꏊ (3) �n�[�h�E�F�A�̒Ǝ҂��������鉷�x�A���x�A�U�����̊����� (4) �d���A�ڒn���̐ݒu���� (5) �ʐM�A���o�͂Ɋւ���d�l (6) �n�[�h�E�F�A�̐ݒu�̓K�ۂ̊m�F���@ (7) �\�t�g�E�F�A�̃C���X�g�[���̓K�ۂ̊m�F���@ (8) ���t���K�i���]���ɂ����锻�� (9) �X�P�W���[�� (10) �ӔC�ҋy�ђS���҂̎��� 5.3.2 ���t���K�i���]���̎��{(1) �n�[�h�E�F�A�̐ݒu �|���ؒS���҂͐��t���K�i���]���v�揑�Ɋ�Â��āA�n�[�h�E�F�A���K�ɐݒu����Ă��邱�Ƃ��m�F���A���̌��ʂ��L�^������̂Ƃ��� �|���ؐӔC�҂́A�n�[�h�E�F�A�̐ݒu�̓K�ۂ肷����̂Ƃ��� (2) �\�t�g�E�F�A�̃C���X�g�[�� �|���ؒS���҂͊�{�\�t�g�E�F�A���܂߁A�K�ɃC���X�g�[������Ă��邱�Ƃ��m�F���A���̌��ʂ��L�^������̂Ƃ��� �|���ؐӔC�҂́A�\�t�g�E�F�A�̃C���X�g�[���̌��ʂ̓K�ۂ肷����̂Ƃ��� 5.3.3 ���t���K�i���]���̕Ɋւ��镶���̍쐬���ؒS���҂́A���t���K�i���]���̕Ɋւ��镶���i�ȉ��u���t���K�i���]�����v�Ƃ����B�j���쐬���A���ؐӔC�҂̏��F����̂Ƃ���B���t���K�i���]�����ɂ́A�����Ƃ��Ď��̎������L�ڂ�����̂Ƃ��� (1) ���t���K�i���]���ɌW�镶���� (2) �]�����ʂƐ����[�u (3) �ӔC�ҋy�ђS���҂̎��� |
5.4�@�^�]���K�i���]���iOQ�j
5.4.1 �^�]���K�i���]���̌v��Ɋւ��镶���̍쐬���ؒS���҂́A�^�]���K�i���]���̌v��Ɋւ��镶���i�ȉ��u�^�]���K�i���]���v�揑�v�Ƃ����B�j���쐬���A���ؐӔC�҂̏��F����̂Ƃ���B�^�]���K�i���]���v�揑�ɂ͌����Ƃ��Ď��̎������L�ڂ�����̂Ƃ���B (1) �^�]���K�i���]���ɌW�镶���� (2) �V�X�e���̉ғ����ɂ�����@�\�̊m�F���@ (3) �^�]���K�i���]���ɂ����锻�� (4) �X�P�W���[�� (5) �ӔC�ҋy�ђS���҂̎��� 5.4.2 �^�]���K�i���]���̎��{(1) ���ؒS���҂́A�^�]���K�i���]���v�揑�Ɋ�Â��ĕ]�������{���A���̌��ʂ��L�^������̂Ƃ��� (2) ���ؐӔC�҂́A�^�]���K�i���]���̌��ʂ̓K�ۂ肷����̂Ƃ��� 5.4.3 �^�]���K�i���]���̕Ɋւ��镶���̍쐬���ؒS���҂́A�^�]���K�i���]���̕Ɋւ��镶���i�ȉ��u�^�]���K�i���]�����v�Ƃ����B�j���쐬���A���ؐӔC�҂̏��F����̂Ƃ���B�^�]���K�i���]�����ɂ́A�����Ƃ��Ď��̎������L�ڂ�����̂Ƃ��� (1) �^�]���K�i���]���ɌW�镶���� (2) �]�����ʂƐ����[�u (3) �ӔC�ҋy�ђS���҂̎��� |
5.5�@���\�K�i���]���iPQ�j
5.5.1 ���\�K�i���]���̌v��Ɋւ��镶���̍쐬���ؒS���҂́A���\�K�i���]���̌v��Ɋւ��镶���i�ȉ��u���\�K�i���]���v�揑�v�Ƃ����B�j���쐬���A���ؐӔC�҂̏��F����̂Ƃ���B���\�K�i���]���v�揑�ɂ͌����Ƃ��Ď��̎������L�ڂ�����̂Ƃ���B (1) ���\�K�i���]���ɌW�镶���� (2) �V�X�e���̉ғ����ɂ�����@�\�y�ѐ��\�̊m�F���@ (3) ���\�K�i���]���ɂ����锻�� (4) �X�P�W���[�� (5) �ӔC�ҋy�ђS���҂̎��� 5.5.2 ���\�K�i���]���̎��{(1) ���ؒS���҂́A���\�K�i���]�����{�v�揑�Ɋ�Â��āA���\�K�i���]�������{���A���̌��ʂ��L�^������̂Ƃ��� (2) ���ؐӔC�҂́A���\�K�i���]���̌��ʂ̓K�ۂ肷����̂Ƃ��� 5.5.3 ���\�K�i���]���̕Ɋւ��镶���̍쐬���ؒS���҂́A���\�K�i���]���̕Ɋւ��镶���i�ȉ��u���\�K�i���]�����v�Ƃ����B�j���쐬���A���ؐӔC�҂̏��F����̂Ƃ���B���\�K�i���]�����ɂ́A�����Ƃ��Ď��̎������L�ڂ�����̂Ƃ��� (1) ���\�K�i���]���ɌW�镶���� (2) �]�����ʂƐ����[�u (3) �ӔC�ҋy�ђS���҂̎��� |
5.6�@�K�i���]���̈ꕔ�ȗ��ƈ��p(1) �u5.4 �^�]���K�i���]��(OQ)�v�ɂ����錟�ؓ��e�A���A�����Ȃǂ��u5.5 ���\�K�i���]���i�o�p�j�v�̓��e�ƍ����Ȃ��ꍇ�͉^�]���K�i���]�����ȗ����Ă������x���Ȃ����̂Ƃ���B�A�����̏ꍇ�A�ȗ��̎|���u�o���f�[�V�����v�揑�v�Ⴕ���́u���\�K�i���]���v�揑�v���͂����ꂩ�̕��ɖ��L���邱�ƁB (2) �e�`�s���͂r�`�s���s�����ꍇ���A���̊m�F�̕��@�y�ыL�^�����ؐӔC�҂ɂ���ēK�ƔF�߂���ꍇ�ɂ́A�K�i���]���ɂ������āA���̌��ʂ����p���Ă������x���Ȃ����̂Ƃ���B |
5.7�@�o���f�[�V�����̑S�̕Ɋւ��镶���̍쐬���ؒS���҂́A�o���f�[�V�����̊e�i�K�̌��ʋy�ё����]�����܂Ƃ߂��o���f�[�V�����̑S�̕Ɋւ��镶�����쐬���A���ؐӔC�҂̏��F����̂Ƃ���B |
�U�D�^�p�Ǘ��Ɩ�
6.�@�^�p�Ǘ��Ɩ�
6.1�@�^�p�Ǘ��ɂ�����ӔC�̐������̔��Ǝғ��̓R���s���[�^���V�X�e���̉^�p�Ǘ��Ɋւ�������ƐӔC���߂���̂Ƃ���B |
6.2�@�^�p�Ǘ��̎菇�Ɋւ��镶���̍쐬�����̔��Ǝғ��̓R���s���[�^���V�X�e���̉^�p�Ǘ��̎菇�Ɋւ��镶���i�ȉ��u�^�p�Ǘ��菇���v�Ƃ����B�j���쐬������̂Ƃ���B�^�p�Ǘ��菇���ɂ͌����Ƃ��Ď��̎������L�ڂ�����̂Ƃ���B�A���A�f�p�o�ȗߖ��͂f�l�o�ȗ߂Ɋւ���菇���Ɋ�Â��Ǘ����s�����ڂɂ��ẮA���̎|���L�ڂ��邱�ƁB (1) �^�p�Ɋւ���ӔC�̐��Ɩ��� �|�g�D �|�^�p�ӔC�� (2) �R���s���[�^���V�X�e���̑���Ɋւ��鎖�� �R���s���[�^���V�X�e���̑���̎菇�Ɋւ��镶���i�W������菇���j���R���s���[�^���V�X�e�����Ƃɍ쐬���A����Ɋ�Â����삷����̂Ƃ���B (3) �ێ�_���Ǘ��Ɋւ��鎖�� �@ ����_������ �A ����_������ �B �ێ�_������Ǝ҂Ɉϑ�����ꍇ�̎挈�ߎ��� (4) �Z�L�����e�B�Ǘ��Ɋւ��鎖�� �@ �f�[�^�̓��́A�C���A�폜���Ɋւ���S���҂̃A�N�Z�X�����̐ݒ�ƕs���A�N�Z�X�h�~�Ɋւ��鎖�� �A ���ʍ\���v�f�̊Ǘ��Ɋւ��鎖�� �B �n�[�h�E�F�A�ݒu�ꏊ�ւ̗��������Ɋւ��鎖�� (5) �o�b�N�A�b�v�E���X�g�A�Ɋւ��鎖�� (6) �ύX�̊Ǘ��Ɋւ��鎖�� �@ �ύX�̌v��A���F�̎菇�Ɋւ��鎖�� �A �ύX�̉e���]���Ɋւ��鎖�� �B ���̑��A�ύX�ɕK�v�Ȏ��� (7) ��E�i�V�X�e���g���u���j�̊Ǘ��Ɋւ��鎖�� �@ ��E�i�V�X�e���g���u���j�������̑Ή��̂��߂̑g�D�� �A ��E�i�V�X�e���g���u��)�̌����̋����y�щe���]�� �B �Ĕ��h�~�����Ɋւ��鎖�� �C �[�u�Ɋւ���菇 �D �V�X�e����~��̍ĊJ�菇�y�эĊJ���̊m�F���� �E ���̑���E�̊Ǘ��ɕK�v�Ȏ��� (8) �S���҂̋���P���Ɋւ��鎖�� (9) ���ȓ_���Ɋւ��鎖�� |
6.3�@�R���s���[�^���V�X�e���̑���̎菇�Ɋւ��镶��(�W������菇��)�̍쐬(1) 6.2(2)�ɋK�肷��W������菇���ɂ͈ȉ��̎������L�ڂ��� �@ �V�X�e���̒S���� �A �R���s���[�^���V�X�e���̑���Ɋւ��鎖�� �B �R���s���[�^���V�X�e���̕ێ�_���Ɋւ��鎖�� �C �R���s���[�^���V�X�e���̃Z�L�����e�B�Ǘ��Ɋւ��鎖�� �D ���̑��A�R���s���[�^���V�X�e���̓����ɉ������^�p�Ǘ��ɕK�v�Ȏ��� (2)�^�p�ӔC�҂́A�W������菇���Ɋ�Â��A�S���҂ɃR���s���[�^���V�X�e���𑀍삳������̂Ƃ���B |
6.4 �ێ�_�������̎��{�^�p�ӔC�҂́A�^�p�Ǘ��菇���Ɋ�Â��A���Ɍf���鎖�����s�����̂Ƃ���B (1) �S���҂ɕێ�_�������{�����A���̌��ʂ��L�^�����邱�ƁB (2) �ێ�_���̋L�^�ɂ��ێ�_���Ǘ����K�ɍs���Ă��邱�Ƃ��m�F���邱�ƁB |
6.5�@�Z�L�����e�B�Ǘ��̎��{�^�p�ӔC�҂́A�^�p�Ǘ��菇���Ɋ�Â��A���Ɍf���鎖�����s�����̂Ƃ���B (1) �f�[�^�̓��́A�C���A�폜���Ɋւ���S���҂̃A�N�Z�X�����̐ݒ�ƁA�s���A�N�Z�X�̖h�~�[�u���u���邱�ƁB (2) ���ʍ\���v�f���̎戵���ɂ��āA�@���ی��}�邱�ƁB (3) �K�v�ɉ����ăn�[�h�E�F�A�ݒu�ꏊ�ւ̗����������s�����ƁB (4) �Z�L�����e�B�Ǘ��Ɋւ���L�^���쐬����ƂƂ��ɁA�����ۊǂ��邱�ƁB |
6.6�@�o�b�N�A�b�v�y�у��X�g�A�^�p�ӔC�҂́A���炩���ߎw�肵���҂ɑ��A�^�p�Ǘ��菇���Ɋ�Â��A���Ɍf����Ɩ����s�킹�邱�ƁB (1) �\�t�g�E�F�A�y�уf�[�^�̃o�b�N�A�b�v���s�����ƁB (2) ��Q��������̉̂��߂Ƀ\�t�g�E�F�A�y�уf�[�^�̃��X�g�A���s�����ƁB (3) �o�b�N�A�b�v�y�у��X�g�A�Ɋւ���L�^���쐬����ƂƂ��ɁA�����ۊǂ��邱�ƁB |
6.7�@�ύX�̊Ǘ��^�p�ӔC�҂́A���炩���ߎw�肵���҂ɑ��A�^�p�Ǘ��菇���Ɋ�Â��A���Ɍf����Ɩ����s�킹�邱�ƁB (1) �ύX���R���s���[�^���V�X�e���ɗ^����e����]�����A�]���̌��ʂɊ�Â��K�ȑ[�u�����{���邱�ƁB�Ȃ��]���̌��ʁA�o���f�[�V�������K�v�Ɣ��f���ꂽ�ꍇ�́A���X�N�̒��x�ɉ����āu�S. �J���Ɩ��v�y�сu�T. ���؋Ɩ��v�ɖ߂��ăo���f�[�V���������{���邱�ƁB (2) �ύX�ɔ�����������菇�Ɋւ��镶���̕ύX�ӏ�����肵�A�K�v�ȉ�������{���邱�ƁB (3) �ύX���e�̊W�҂ւ̎��m�̕��@�����肵�A�K�v�ɉ����ċ���P�������{���邱�ƁB (4) �ύX�̊Ǘ��̋L�^���쐬���A�^�p�ӔC�҂̊m�F��ƂƂ��ɁA�ύX�̊Ǘ��Ɋւ���ӔC�ғ��̏��F�邱�ƁB �f�l�o�ȗ߂ɌW��V�X�e���Ɋւ���ύX�̊Ǘ��ɂ��ẮA�f�l�o�ȗ߂ɂ�����ύX�̊Ǘ��̎菇�ɏ]���ĉ^�p���邱�ƁB�������A���̏ꍇ����L(1)����(4)�̓��e���܂ނ��ƁB |
6.8�@��E(�V�X�e���g���u��)�̊Ǘ��^�p�ӔC�҂́A���炩���ߎw�肵���҂ɑ��A�^�p�Ǘ��菇���Ɋ�Â��A���Ɍf����Ɩ����s�킹�邱�ƁB (1) ����������E�i�V�X�e���g���u���j�����i�̕i���ɋy�ڂ��e����]�����A���₩�ɓK�ȑΉ��[�u���u����ƂƂ��ɁA���̌������������A�K�v�ȍĔ��h�~�[�u�����{���邱�ƁB (2) ��E�i�V�X�e���g���u���j������ɃR���s���[�^���V�X�e���̉^�p���ĊJ����ꍇ�ɂ́A���A�ғ����K�ɍs���Ă��邱�Ƃ��m�F���邱�ƁB (3) ��E�i�V�X�e���g���u���j�̊Ǘ��̋L�^���쐬���A�^�p�ӔC�҂̊m�F��ƂƂ��ɁA��E�̊Ǘ��Ɋւ���ӔC�ғ��̏��F�邱�ƁB �f�l�o�ȗ߂ɌW��V�X�e���Ɋւ����E�̊Ǘ��ɂ��ẮA�f�l�o�ȗ߂ɂ������E�̊Ǘ��̎菇�ɏ]���ĉ^�p���邱�Ƃł悢���A���̏ꍇ����L(1)����(3)�̓��e���܂ނ��ƁB |
6.9�@����P��
6.9.1 ����P���v��̍쐬�^�p�ӔC�҂́A�^�p�Ǘ��菇���Ɋ�Â��A���炩���ߎw�肵���҂ɁA�R���s���[�^���V�X�e�����g�p�����Ɩ��ɏ]������҂ɑ��鋳��P���v����쐬�����邱�ƁB�Ȃ��A����P���ɂ��Ă͂f�p�o�ȗ߁A�f�l�o�ȗ߂ɂ�����菇�ɏ]���ĉ^�p���邱�Ƃ��]�܂����B 6.9.2 ����P���̎��{�^�p�ӔC�҂́A����P���v��Ɋ�Â��A���炩���ߎw�肵���҂Ɏ��Ɍf����Ɩ����s�킹�邱�ƁB (1) �R���s���[�^���g�p�����Ɩ��ɏ]������҂ɑ��āA�R���s���[�^���V�X�e�����g�p�����Ɩ��Ɋւ��鋳��P�����v��I�Ɏ��{���A���̋L�^���쐬���邱�ƁB (2) ����P���̎��{�ɂ��ĉ^�p�ӔC�҂̊m�F��ƂƂ��ɁA�i���ۏؐӔC�Җ��͐����Ǘ��ҎႵ���͐ӔC�Z�p�҂ɑ��ĕ����ɂ����邱�ƁB 6.9.3 ����P���̋L�^�̕ۊ��^�p�ӔC�҂͋���P���̎��{�̋L�^��ۊǂ��邱�ƁB |
�V�D���ȓ_��
7 ���ȓ_��
7.1 ���ȓ_���̎��{�����̔��Ǝғ��́A�菇�����Ɋ�Â��A���炩���ߎw�肵���҂Ɏ��Ɍf����Ɩ����s�킹�邱�ƁB�Ȃ��A���ȓ_���ɂ����ẮA�f�p�o�ȗ߁A�f�l�o�ȗ߂ɂ�����菇�ɏ]���ĉ^�p���邱�Ƃ��]�܂����B (1) �R���s���[�^���V�X�e�������̃K�C�h���C���Ɋ�Â��A�^�p�Ǘ�����Ă��邱�Ƃ��m�F���邽�߂ɒ���I�Ɏ��ȓ_�������{���邱�ƁB (2) ���ȓ_���̌��ʂɂ��ĕi���ۏؐӔC�Җ��͐����Ǘ��ҎႵ���͐ӔC�Z�p�҂ɑ��ĕ����ɂ����邱�ƁB (3) ���ȓ_���̌��ʂ̋L�^���쐬���A�����ۊǂ��邱�ƁB 7.2 ���P�[�u�̎��{ �����̔��Ǝғ��́A���ȓ_���̌��ʂɊ�Â��A���P���K�v�ȏꍇ�ɂ́A���炩���ߎw�肵���҂ɏ��v�̑[�u���u�������A���̋L�^���쐬���K�Ȏ҂ɕ�����ƂƂ��ɁA�����ۊǂ����邱�ƁB |
�W�D�R���s���[�^�V�X�e���̔p��
8.�@�R���s���[�^�V�X�e���̔p��
8.1 �R���s���[�^�V�X�e���̔p���̌v��Ɋւ��镶���̍쐬�����̔��Ǝғ��̓R���s���[�^�V�X�e���̔p���ɂ������ẮA�R���s���[�^�V�X�e���̎�ނ�K�́A�J�e�S�����A�K�v�ɉ����āA�R���s���[�^�V�X�e���̔p���Ɋւ���v�揑�i�ȉ��u�p���v�揑�v�Ƃ����B�j���쐬���邱�ƁB�p���v�揑�ɂ͌����Ƃ��Ĉȉ��̎������L�ڂ�����̂Ƃ���B (1) �p���Ɋւ���ӔC�̐��Ɩ��� �|�g�D �|�R���s���[�^�V�X�e���̔p���̐ӔC�� (2) �p���ΏۂƂ���R���s���[�^�V�X�e�� (3) �f�[�^�̈ڍs�Ɋւ��鎖�� (4) �Z�L�����e�B�Ɋւ��鎖�� (5) �R���s���[�^�V�X�e���̔p�����@ �R���s���[�^�V�X�e���̎�ނ�K�́A�p�r���ɉ����Ĉȉ����Q�l�ɂ��ēK�ɒ�߂邱�� �|���X�N�A�Z�X�����g �|�O����� �|�X�P�W���[�� �|��̓I�Ȕp���̕��@ �E�n�[�h�E�F�A �E�\�t�g�E�F�A �E�f�[�^ �E�����ށi�菇���A�L�^�A�_���j (6) �p�������̔��f� |
8.2�@�R���s���[�^�V�X�e���̔p���L�^�̍쐬�R���s���[�^�V�X�e���̔p���̐ӔC�҂́A�p���v�揑�Ɋ�Â��R���s���[�^�V�X�e����p������ƂƂ��ɁA�p���̋L�^���쐬���A�����ۊǂ��邱�ƁB |
�X�D�����y�ыL�^�̊Ǘ�
9.�@�����y�ыL�^�̊Ǘ����̃K�C�h���C���Ɋ�Â��쐬���ꂽ�����y�ыL�^�͂f�p�o�ȗߖ��͂f�l�o�ȗ߂Ɋ�Â���߂������y�ыL�^�̊Ǘ��̕��@�ɏ]���ēK�ɕۑ��Ǘ�������̂Ƃ���B�f�p�o�ȗߋy�тf�l�o�ȗ߂ɂ܂�����V�X�e���̏ꍇ�́A���炩���߂ǂ���̏ȗ߂ɏ]���ĊǗ����邩���^�p�Ǘ��菇���ɖ��L���Ă������ƁB |
�P�O�D�p��W
10. �p��W
�^�]���K�i���]���i�n�p�j�R���s���[�^���V�X�e�����A�ғ����Ɠ����̊����ɂ����āA�@�\�d�l�Ɏ����ꂽ�^�]�͈͂ňӐ}�����悤�ɋ@�\�Ɛ��\�����邱�Ƃ��m�F�����������邱�ƁB �^�p�Ǘ��Ɩ��R���s���[�^���V�X�e���̉ғ��J�n��A�R���s���[�^���V�X�e�����A�o���f�[�g���ꂽ��Ԃ��ێ����A�v���d�l�ɋL�ڂ��ꂽ�v���Ɋ�Â��ēK���ɉғ������邽�߂̋Ɩ��B �^�p�ӔC���R���s���[�^���V�X�e���^�p�Ɩ����s�����߂̐ӔC�҂Ƃ��āA�����̔��Ǝғ��ɂ��^�p�Ǘ��菇���ɂ����Ďw�肳�ꂽ�҂������B �J���Ɩ��w�肳�ꂽ�R���s���[�^���V�X�e���̌v��A�v�A����A�e�X�g�A��������܂ł̋Ɩ��B �J���v�揑�w�肳�ꂽ�R���s���[�^���V�X�e�����J������ۂɖړI�A�����A�ӔC�A�̐��A�X�P�W���[���Ȃǂ��L�q���������B �J���ӔC���R���s���[�^���V�X�e���J���Ɩ����s�����߂̐ӔC�҂Ƃ��āA�����̔��Ǝғ��ɂ��J���v�揑�ɂ����Ă��炩���ߎw�肳�ꂽ�҂������B �@�\�d�l���i�e�r�j�v���d�l���ɋL�ڂ��ꂽ�v���d�l�ɑΉ�����A����̓I�ȋ@�\���L�ڂ��ꂽ�����B �������R���s���[�^���V�X�e�����J�����邢�͓������A�����̔��Ǝғ��ɒ���҂������B��ʂɃT�v���C����x���_�[�ƌĂ��B���ЂŊJ������ꍇ�͎��Ђ̃V�X�e���J���҂��܂ށB �����҃A�Z�X�����g�����̔��Ǝғ��ɂ�鋟���҂̑I�莞����J���̊e�i�K�ɂ����鋟���҂̕]���̂��ƁB �����Ҋč������҂̕i���Ǘ��̐���i���ۏ̃V�X�e���A���邢�͌o���E�\�͂���тȂǑ��p�I�ɋ����҂̒������s���A�����҂̑����I�ȕi���}�l�W�����g�V�X�e����\�͂�]���E�m�F���邱�ƁB���n���͏��ʂɂ��č����@������B ���؋Ɩ��R���s���[�^���V�X�e�����A�v���d�l���ɒ�߂��v���ɍ��v���Đv����A�����t�����A�V�X�e���̉ғ����y�щғ���Ԃɂ����āA�@�\�y�ѐ��\�����邱�Ƃ��m�F���邱�ƁB ���ؐӔC�����؋Ɩ����s�����߂̐ӔC�҂Ƃ��āA�����̔��Ǝғ��ɂ��J���v�揑�ɂ����Ă��炩���ߎw�肳�ꂽ�҂������B ���n��������i�r�`�s, Site Acceptance Test�j�����҂��V�X�e�������n�̉ғ����ŋ@�\�y�ѐ��\�̑S�Ă��邢�͈ꕔ���@�\�d�l�����Ă��邱�Ƃ��m�F���邱�ƁB�����ł����u���n�v�Ƃ́A�����̔��Ǝғ������Y�̃V�X�e����ݒu����\��̏ꏊ�������B �H��o�����i�e�`�s, Factory Acceptance Test�j�����҂��V�X�e�����o�ׂ���O�ɐ�����ŋ@�\�y�ѐ��\�̑S�Ă��邢�͈ꕔ���@�\�d�l�����Ă��邱�Ƃ��m�F���邱�ƁB �\���ݒ��R���s���[�^�V�X�e���𗘗p����ɂ������ăn�[�h�E�F�A�y�у\�t�g�E�F�A�̍\���v�f�̑g�ݍ��킹��ғ���������ݒ肷�邱�ƁB���Ȃ킿�A�n�[�h�E�F�A�ɂ����ẮA�V�X�e�����\������A�R���s���[�^�A���Ӌ@�킠�邢�͂����ɑg�ݍ��܂�镔�i�i�{�[�h���j�̑g�ݍ��킹��ݒ肵�A�o�^���邱�ƁB�\�t�g�E�F�A�ɂ����ẮA�v���O�������쐬�A�ύX���邱�ƂȂ��A�V�X�e�����\�����郂�W���[���̑g�ݍ��킹�A�y�уV�X�e�����ғ���������A�p�����[�^����ݒ肵�A�o�^���邱�ƁB �R���s���[�^�V�X�e������̋@�\���͈�A�̋@�\�����s���邽�߂ɁA�v���A�g�ݗ��Ă�ꂽ�n�[�h�E�F�A�y�ъ֘A����\�t�g�E�F�A�̃O���[�v�B �R���s���[�^���V�X�e���R���s���[�^�V�X�e���œ������ꂽ�H�����͍��(����GMP)�A�y�уR���s���[�^�V�X�e���ɂ����������@�\�𗘗p����Ɩ��v���Z�X�B �R���s���[�^�V�X�e���̔p���̐ӔC���R���s���[�^�V�X�e���̔p�����s�����߂̐ӔC�҂Ƃ��āA�����̔��Ǝғ��ɂ��p���v�揑�ɂ����Ďw�肳�ꂽ�҂������B ���ʍ\���v�f�V�X�e���̉^�p�ɂ����đ���҂����ʁA���肷�邽�߂ɗp������f�[�^�̑g�ݍ��킹�A�������͋@��ƃf�[�^�̑g�ݍ��킹�A�Ⴆ��ID�ƃp�X���[�h�̑g�����B �V�X�e���A�Z�X�����g�J���ΏۂƂ���R���s���[�^���V�X�e���̃o���f�[�V�����ɂ����錟�ؓ��e��쐬�����������肷�邽�߂ɁA�V�X�e���̃\�t�g�E�F�A�̕��G����J�����@�A���Y�V�X�e���ɂ�萻������鐻�i�̈��S����i���ւ̉e���̓x�����A���͓��Y�V�X�e���ɂ��쐬�A�ۑ������d�q�L�^�̏d�v�x�A�����҂̃V�X�e���J���ߒ��ł̕i���ۏ̏��𑍍��I�ɕ]�����邱�ƁB �V�X�e���e�X�g�ғ����邽�߂Ɍ������ꂽ��ԂŃ��W���[���A�v���O�������@�\�d�l���A�v�d�l���ʂ�ɋ@�\���邱�Ƃ��m�F���邱�ƁB ���t���K�i���]���i�h�p�j�V�X�e�����A�v���d�l���ɋL�ڂ��ꂽ�Ƃ���ɐ����t�����A�v���O�������C���X�g�[�����ꂽ���Ƃ��m�F���A���������邱�ƁB ���\�K�i���]���i�o�p�j�R���s���[�^���V�X�e�����A�ғ����ɂ����āA�v���d�l�ʂ�ɋ@�\���A���\�����ĉ^�]�ł��邱�Ƃ��m�F���A���������邱�Ƃ������B �v�d�l���i�c�r�j�@�\�d�l�ɋL�ڂ��ꂽ��̓I�ȋ@�\����������R���s���[�^���V�X�e�����쐬���邽�߂̏ڍd�l���L�ڂ��ꂽ�����ŁA�n�[�h�E�F�A�d�l���ƃ\�t�g�E�F�A�d�l���ɕ�������ꍇ������B �n�[�h�E�F�A�d�l���F�V�X�e�����\������n�[�h�E�F�A�̎d�l�A�\�����L�q���� �\�t�g�E�F�A�d�l���F�V�X�e�����\������\�t�g�E�F�A�̏ڍ@�\�A�\�����L�q���� �v���K�i���]���i�c�p�j�v���d�l���ɋL�ڂ��ꂽ�v���������A�ȍ~�̋@�\�d�l���A�v�d�l���ɐ��������f����Ă��邱�Ƃ��m�F�����������邱�ƁB �\�t�g�E�F�A�J�e�S�������x�̐M������L����\�t�g�E�F�A�̑����ׂ��͈́B�\�t�g�E�F�A�̐�����������敪�����ł̊�{�I�ȕ��ށB �v���O�����d�l���v�d�l���̋@�\���������邽�߂Ƀ��W���[���A�v���O�����Ŏ������ׂ��������L�q�����d�l���B �v���O�����e�X�g���W���[���A�v���O�������P�̂Ńv���O�����d�l���ʂ�ɋ@�\���邱�Ƃ��m�F���邱�ƁB ���W���[���\�t�g�E�F�A���\������@�\�̍ŏ��P�ʁB �v���d�l��(�t�q�r)�w�肳�ꂽ�R���s���[�^���V�X�e���Ɋւ���@�\��̗v���d�l���L�ڂ��ꂽ�����B ���X�N�A�Z�X�����g���X�N�}�l�W�����g�v���Z�X�̒��ŁA���X�N�ɌW��錈����x�������������n���������v���Z�X�B�n�U�[�h�̓���A�y�т����n�U�[�h�ւ̔��I�ɔ������X�N�̕��͂ƕ]������Ȃ�B ���X�g�A���炩���ߓK�Ȕ}�̂Ƀo�b�N�A�b�v���Ă������A�v���O�����A�p�����[�^�A�f�[�^�����A�ēx�V�X�e���ɓǂݍ��܂��A�V�X�e�����o�b�N�A�b�v�������_�Ɠ��l�̏�Ԃɖ߂����ƁB ���̑��̗p��ɂ��ẮA�f�p�o�ȗ߁A�f�l�o�ȗߋy�ъ֘A�̒ʒm�ނ̗p����Q�ƁB |
�ʎ�1�@�R���s���[�^���V�X�e���̃��C�t�T�C�N�����f��
�ʎ�2�@�J�e�S�����ޕ\
�Q�l
- ���i�E��O�i�����̔��Ǝғ��ɂ�����R�� �s���[�^���V�X�e���K���Ǘ��K�C�h���C���i�āj�@�����J���ȁ@2010�N7��16��


