
1. はじめに
前号でも紹介したとおり、日本製薬工業協会 医薬品評価委員会は、2007年11月1日、臨床試験データを電子的に取得する場合の具体的な要件を示すことを目的に「臨床試験データの電子的取得に関するガイダンス」(以下、ガイダンス)と呼ばれる自主ガイダンスを発行した。
症例報告書(以下、CRF)を電子化する場合、これまでの紙媒体のCRF(以下、紙CRF)と同等の品質および品質保証を確保する必要がある。
EDCは治験の生データである症例データを電子的に取得するものであり、ERESガイドラインの適用を受ける。
ガイダンスは、ERESガイドラインを具体的にEDCに適用させたものである。
しかしながらガイダンスは、いくつかの用語が定義されないまま使用されていたり、本来の用語の意味と多少解釈の違う点が見られる。また真正性、見読性、保存性の区別が多少入り乱れている個所も見受けられる。
本稿では、ガイダンスの内容を筆者なりに考察してみたい。
2. 電子症例報告書を原本とすることができる要件
ガイダンスの第4章は、「臨床試験データを電子的に取得するための要件」であり、「4.1. 実施医療機関で入力されるデータについての要件」と「4.2. 中央検査機関から電子的に入手するデータについての要件」に分かれている。
4. 臨床試験データを電子的に取得するための要件 |
このセクションを読む限り、本ガイダンスは、電子症例報告書(以下、eCRF)を原本とすることができる要件を記載していると読める。しかしながら、これまでのように紙CRFをEDCシステムを利用して作成する場合にも適用するべきであると考える。
電子症例報告書原本は、ASPサービス利用中(つまり治験実施中)は、サーバー上のデータベースにあり、ASPサービス終了後(つまり治験終了後)にはCD-R等の電磁的記録媒体にpdf等のフォーマットで出力され管理される。(図1 参照)
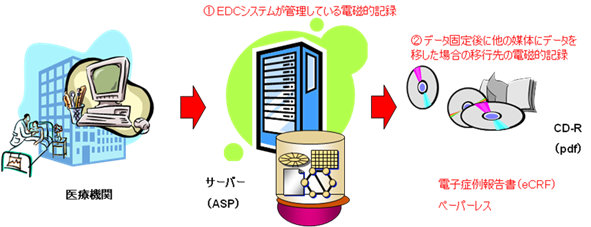
図1 電子症例報告書原本
ただし、電子症例報告書原本は、EDCシステム稼動中及びEDC サーバーからのデータ移管後の各段階で、予め定義・特定されていなければならない。 |
規制当局は、いかなる時点においても臨床試験データを査察することがあるので、EDC利用に際しては、時系列的にどの電磁的記録が「電子症例報告書原本」であるかをプロトコール(またはSOP)で定義しておく必要があるのである。
3. コンピュータシステムバリデーション
なお、使用するEDCシステムはコンピュータシステムバリデーション(Computerized System Validation 以下CSV)ポリシーに則ったCSVによりシステムの信頼性が保証されていることが前提であり、以下の点に留意する。 |
「CSVポリシー」とは、製薬企業各社が作成しているCSVに関する手順書等の文書のことであると理解する。
現在、日本の規制当局からはCSVに関するガイドラインが出されていない。
多くの製薬企業では、GAMP等に従って、CSVの手順書等を作成しているものと思われる。
EDCシステムをASPサービスとして利用する場合、当該システムの品質保証は、その多くを当該ベンダーが実施しているべきである。ベンダーの多くは自社のQMS(品質管理システム)を文書化しているはずであり、それらに則った品質保証が実施されていることを期待する。
EDCを利用する製薬企業は、当該ベンダーを事前に監査しておかなければならない。またユーザ受入れテスト(UAT)を実施し、要求仕様を満たすことを確認しておく必要がある。
|
このパラグラフは、CSV SOPの作成に関する注意事項であると読める。
システムの運用段階で重要なことは、当該システムがバリデーション状態を維持していることを保証することである。そのためには、運用段階における障害管理及び変更管理の手順を定めておかなければならない。
システムの改訂とは、当該ソフトウェア等のバージョンアップ等を指すと考えられる。改訂も変更管理に従って実施しなければならない。
システムの廃棄段階で重要なことは、 当該症例データのみならず、監査証跡、電子署名の情報を別のシステム等に移行しておかなければならない。
CSV実施で重要なことは、文書化された証拠つまり記録を作成し適切に保存しておくことである。
|
規制当局の査察等が行われた際には、CSV SOP等を提示し、説明できることが肝心である。
CSV報告書とは、CSV SOP等に従って作成したCSVの実施記録のことであると理解する。CSVの記録は適切に保存しておかなければならない。これら必要な資料は、すみやかに提示できるようにしておかなければならない。
|
EDCベンダーやASPベンダーは、当該システムの品質保証を行う義務がある。またCRO等は、品質が保証されたEDCシステムを利用し、その治験実施のプロセスの品質を保証し、また当該治験における症例データ(電磁的記録)等の品質を保証する義務がある。
製薬企業は、当該EDCシステムについて、それらベンダーが自社のQMS等に従って品質保証していることを、ベンダーオーディット等を通じて事前に確認しておかなければならない。
また治験実施中においても、CRO等が品質計画を守って活動し、症例データが適正に取得され保存されていることを常にモニタリングしなければならない。
ベンダーやCRO等に対して、ベンダーオーディット等を実施する際には、本ガイドラインに従ってチェックリスト等を作成し、要件を満たすかどうかを調査しておくことが望まれる。
4. 電磁的記録の真正性に関する要件
4.1.1. 電磁的記録の真正性に関する要件
|
真正性とは、当該記録が本物であり、記録の作成者自身が入力または修正しており、主張通りの時刻に作成されていることを言う。
真正性を確保するためには、セキュリティや監査証跡等の機能要件と、当該システムをルールに従って適正に利用するといった運用要件の両方を検討しなければならない。
ユーザを管理する際には、あらかじめ決められたルールに従って、権限設定を適切に行わなければならない。権限設定とは、入力権限、修正権限、削除権限、承認権限等である。
権限は必要なユーザに最低限で設定するべきで、不要になった際にはすみやかに抹消しなければならない。
パスワードは決して他人に漏らしてはならない。システムがセキュリティ機能を持っていても、運用する者がルールを守らない限り、その信頼性は確保できない。
|
治験責任医師、分担医師、治験協力者等へ、ID、パスワード等を適切に運用するよう、教育研修を行い周知徹底しておかなければならない。
なりすましとは、本人ではないものがパスワードを盗用して、あたかも本人であるかのように入力・修正等を行ってしまうことである。
教育研修において、EDC運用中にセキュリティを侵害するような事態があった場合には、すみやかに報告義務があることなどを説明しておかなければならない。
また教育研修を実施したことをモニタリング報告書等で記録し、証拠としておくことが望ましい。
|
上記の機能要件は、事前(すなわちASP契約前)にチェックしておかなければならない。
またCSVを徹底し、上記要件を検証しておかなければならない。
監査証跡は、規制当局等が改ざんの有無を調査するために必要で、自動的に記録されなければならない。
監査証跡自体が改変されれば、電磁的記録の真正性が確保できないことになる。規制当局にとって、監査証跡は“最後の砦”である。
|
信頼性・品質保証とあるが、信頼性は品質保証の一部である。
ちなみに品質保証は英語でQuality Assuranceであり、信頼性はReliabilityという。Integrity(完全性)も品質保証上、重要な要件である。
2) EDCシステム及び運用手順において、セキュリティが確保されている。
|
これら機能要件においても、事前(すなわちASP契約前)にチェックし、CSVを徹底しておかなければならない。
|
インターネット等を利用し、複数のサーバーを経由している途中で、データを盗み見られたり、差し替えられないように、SSL(セキュア・ソケット・レイヤー)や、VPN(仮想プライベートネットワーク )のような暗号化通信技術の利用が必須である。
3) GCPの下、紙症例報告書を利用して臨床試験データを収集した場合と同じレベルの品質が、電子症例報告書作成プロセスの運用、管理により確保されている。
|
電子化の原則は、紙ベースのオペレーションが電子化された際に、データの品質および品質保証が劣化してはならないということである。
|
電子症例報告書が原資料となるデータとは、カルテ等に記載しないデータのことである。紙CRFでも同様であるが、あらかじめプロトコール等に記載しておかなければならない。
電子症例報告書が原資料となるデータの場合、SDV等で信頼性を保証する手立てがない。
「システムでの入力制御を運用手順で補完する場合」とあるが、おそらく権限設定機能が不十分なEDCシステムを指しているものと思われる。そのようなシステムを利用することは控えるべきである。
|
GCPでは、治験責任医師が自ら症例報告書の写しを作成し、原本を治験依頼者に提出することになっている。
EDCを利用した治験の場合は、eCRFを治験依頼者が先に入手することになるので、製薬会社が写しを作成し、治験責任医師に提供することとなる。
具体的には、CD-R等のメディアにeCRFをpdf形式でコピーし提供することになる。
pdf形式を利用する理由は、見読性を確保するためである。
CD-R等のメディアは、経年劣化するという問題がある。紙CRFの場合は、ほぼ永久に保存できる。しかしながら、CD-Rは、直射日光をあてたり、割ったり、高温多湿状態で保管した場合、読みだすことができなくなる危険性がある。
したがって、医療機関に対し、保存性確保のための手順書(取り扱い方、保存環境等)を交付しておくことが望ましい。
さらにCD-Rの破損時の交換が可能なように、コピーを依頼者側でも保管しておかなければならない。
■治験責任医師が保存する電子症例報告書の写しは、以下の要件を満たしている。
|
eCRFの写しを作成する際は、手作業をはさまず、無変換又は検証された自動変換の方式により出力されなければならない。これは写しが正確であることを保証するためである。
「予め定められた保存期間中」とは、GCP上の必須文書の保存期間を指すものと思われる。
規制当局が調査できるためには、ドライブと当該バージョンを読み出せるソフトウェアが医療機関に必要となる。
症例報告書の作成の記録及び治験責任医師の署名については、以下のように運用されている。
|
アカウント管理表は、署名・印影一覧表に相当する。(図2 参照)
アカウント管理表は、適宜更新する必要がある。ただし、何らかの事情でユーザでなくなった場合でも、ユーザ登録を削除してはならない。その場合は、アクセス権限を削除し、ログオンができないようにする必要がある。
| 医療機関名 |
職位 |
氏名 |
アカウント |
eMail/TEL |
権限 |
備考 |
図2 アカウント管理表
GCP 第47 条第1 項及び第2 項で症例報告書作成、変更又は修正の際に必要とされている記名捺印又は署名に関して、EDC を用いる場合は以下のとおりに対応する。
|
すべてのデータは、入力者および入力日時が監査証跡等で特定できなければならない。そのような機能をもったEDCを選択する必要がある。多くの場合は、CRCが入力することになると思われる。
ビジット毎の症例票に対して、治験責任医師または治験分担医師が電子署名を付すかどうかは、各社のSOPによって異なる。
症例毎にロジカルチェックが終了し、すべてのクエリーが解決され、コーディングが完了すれば、治験責任医師が電子署名を行うことになる。
紙CRFを原本とする場合は、治験責任医師は、印刷後すみやかに手書き署名または記名・捺印を行う必要がある。
|
電磁的記録を紙に印刷し、紙CRFに手書き署名または記名・捺印を行うといった、ハイブリッドシステムの場合、報告書と対応する署名(記名・捺印を含む)のリンクは困難である。
治験責任医師はeCRF、紙CRFともに、署名(記名・捺印を含む)を行う際は監査証跡を確認し、改ざん(意図しない変更)が行われていないことを確認して行わなければならない。
紙CRFには、監査証跡の情報が印刷されるEDCを選択しておくことが望ましい。
また監査証跡確認手順を、操作マニュアル等に記載しておく必要がある。
5. おわりに
EDCに見られるように、電子化の波は何人も止めることはできない。
21 CFR Part 11やERESガイドラインは、いわば規制緩和である。これまで書面で保存が義務つけられていた記録を、電磁的記録で保存しても良いとしたからである。
これまで電磁的記録は紙媒体の記録に比べて、データの品質および品質保証を劣化させてはならないとされてきた。しかしながら適切にバリデートされたコンピュータシステムは、紙媒体による記録よりも信頼性が高いといえるのである。
次号も引き続き、ガイダンスの考察を続けたい。
参考
- 「医薬品等の承認又は許可等に係る申請等における電磁的記録及び電子署名の利用について」平成17年4月1日 薬食発第0401022号
- 「臨床試験データの電子的取得に関するガイダンス」平成19年11月1日 日本製薬工業協会 医薬品評価委員会


