PIC/S GMP における品質保証の留意点

ウェブセミナー
PIC/Sについて研究するページです。*万が一文中に解釈の間違い等がありましても、当社では責任をとりかねます。
本文書の改訂は予告なく行われることがあります。
PIC/S GMP における品質保証の留意点
はじめに
手前味噌な話だが、筆者はしばしばサプライヤオーディット(供給者監査)を支援している。
我々コンサルタントは、プロであるため、必ず何らかの指摘事項を発見し、また適切な改善勧告を行わなければならない。
先日、こういうサプライヤ(ITベンダー)があった。これまでに通算17社の監査を受けてきたが、まだ一度もメジャーな指摘を受けたことがないとのことであった。
念のため、これまでの監査実施企業名の提示を求めたところ、国産大手企業や外資系企業なども含まれていた。
しかしである。筆者が監査を行ってみると、いとも簡単に指摘事項が見つかった。
どういうことかというと、当該サプライヤの某プロジェクトマネージャは、完璧なまでにプロジェクト文書を作成し、またテストも十分に実施していた。それらの記録も完備していた。プログラムテスト、単体テスト、連結テスト、インテグレーションテストといった具合である。
さすがに17社の監査で指摘がなかったはずである。
しかしである。筆者が、ではどのSOPに則ってそれら記録を作成したのかを尋ね、該当するSOPを見せてもらったところ、当該SOPにはテストに関する詳細な手順の記載がなかった。
つまり、当該プロジェクトマネージャは、自身の経験とスキルで当該プロジェクトを遂行していたのである。
何が問題であるかお分かりであろうか。
我々は、ともすると結果が良ければそれで良いと思いがちである。しかしながら、上述の通り、当該プロジェクトマネージャはSOPを拠り所としていなかった。これはSOP違反である。つまりSOPに書いていないことを実施していたわけである。
ただし、お分かりのように、悪いのはSOPの方である。当該プロジェクトは、優秀なプロジェクトマネージャが担当したので、良い結果となったが、もし別の担当者だった場合、どのような結果となっていただろうか。SOPが不十分であるので、当然のことながら品質の十分ではない結果になったであろう。
これが筆者の観察結果である。
規制要件とポリシー
読者各位に質問したい。規制要件はいったい誰に向けて発出されているかご存知であろうか。
正解は、各企業のトップ(CEO)宛に出されているのである。つまり、製薬企業の従業員に直接出されているものではないのである。
企業のトップは、規制要件を受けて、自社のポリシー(規程、規定)を作成しなければならない。
また規制要件の数だけ、ポリシーが作成されなければならない。例えば、Part11対応ポリシー、コンピュータ化システム適正管理ガイドライン対応ポリシー(コンピュータ化システム管理規定)、ER/ES指針対応ポリシーなどである。
ではなぜ、同じ規制要件に対して、各社で異なるポリシーが必要かというと、各社によって生産している製品(薬剤)が異なるためリスクが違ったり、従事するプロセスが異なったり、規模が異なったりするためである。
各企業のトップは、自社の製品、リスク、プロセス、規模などに適切に合致させた規制要件対応ポリシーを作成しなければならないのである。
従業員は、トップが決めたポリシーを遵守しなければならないのである。また自社のポリシーに従った手順書(SOP)等を作成しなければならない。
面白いエピソードがある。ある外資系企業の監査を受けた日本の製薬企業での話である。監査担当者が、開口一番、「ポリシーを見せて欲しい。」といったそうである。しかしながら、その企業はポリシーと呼ばれる文書を作成していなかったため、「ポリシーはありません。」と答えてしまったそうである。
外国人から見ると、ポリシーがないというのは、非常に奇妙(付き合っていけるか不安)な企業である。
当該担当者が言いたかったことは、ポリシーと呼ばれる文書は作成していないが、手順書(SOP)はちゃんと整備しているということだったのだろう。
おおむね、我々日本人は、ポリシーという文書を作成するのが苦手である。
これも手前味噌であるが、筆者は会社を経営しており、採用面接を行うことがしばしばある。その際に「あなたのポリシーは何ですか?」という質問をよく行うことがある。しかしながら、たいていの日本人は答えに窮してしまう。つまり日本人や日本企業では、ポリシーを考えることが苦手である。
企業のトップは、自社のポリシーを定め、従業員のすべてが、そのポリシーを遵守するように徹底させなければならない。このことを内部統制(Corporate Gavernance)と呼ぶ。
トップがポリシーを作成せず、現場任せであっては、ガバナンスが効いている状態とは言えない。
ISO-9000は2000年版から、企業の経営者が品質責任を持つ形に大きく変更となった。これまでは、従来の企業経営者は、デシジョン(意思決定)を行うために、経済的な事情を優先してきた。つまり、損得勘定である。しかしながら、品質問題があちらこちらで発生している現状では、企業のトップは、金銭面だけで判断してはならないということになったからである。
例えば、毎年冬になるとPanasonic(旧松下電器)が、石油ファンヒータの回収をテレビコマーシャルなどで呼びかけている。その費用は莫大なものであろうと推察する。しかしながら、費用よりも品質(安全)を優先するという姿勢は、非常に素晴らしいことである。これによりPanasonicは、企業のブランド力を高めたことに違いない。
日本では2010年に発出された、ICH Q10 品質システム(QS)においても、同様に製薬企業のトップの品質責任を重要視している。
PIC/S GMPにおける品質保証
これまでのGMPは、製造基準が中心であった。FDAは、21世紀のcGMPのイニシャティブの最終報告の中で、cGMPの要件の拡大をあげている。追加の要件とは、品質保証と品質リスクマネージメントである。
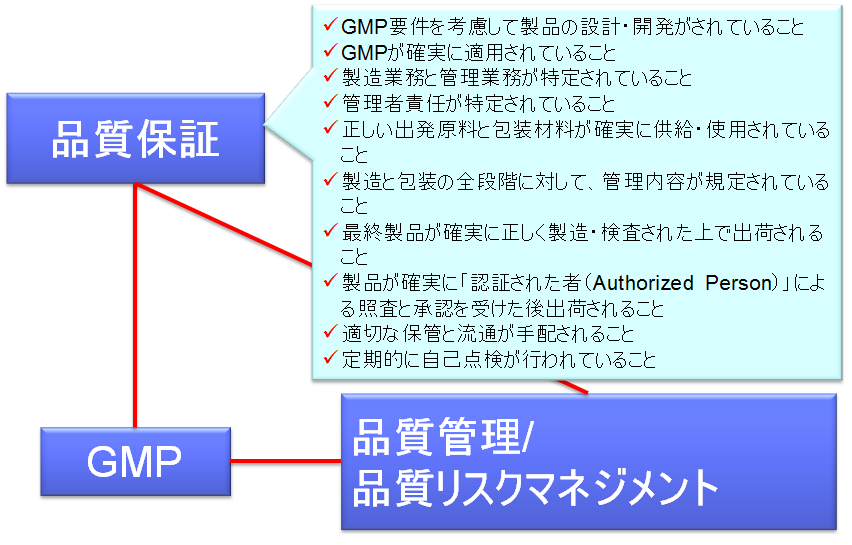
PIC/S GMPの有機的な繋がり
このFDAの提唱は、ICHでも議論され、品質に関するガイドラインとして、いわゆるQトリオが三極で合意された。
Qトリオとは、
- Q8 製剤開発に関するガイドライン
- Q9 品質リスクマネジメントに関するガイドライン
- Q10 医薬品品質システム
であり、本邦でもすべて通知(Step 5)されている。
この中で、Q8は製剤における品質保証のみを対象としているのに対して、Q10は製薬企業全体の品質保証を求めている。
その適用範囲は非常に広範囲で、原材料の購入からディストリビューションにまで及ぶ。
サプライヤオーディットによる品質保証
原材料を購入する際には、経理スタッフではなく、技術スタッフの関与が必要で、定期的なサプライヤオーディットが求められる。もちろんコンピュータ化システムを導入する際にもサプライヤオーディットは必須である。
GDP(Good Distribution Practice)
また、ディストリビューションは、本邦においては卸業者が適切に医薬品の流通を執り行っているが、諸外国−とりわけ後進国などでは、流通の過程で搬送トラックごと盗難に遭うことすら発生しているという。
盗難に遭うことよりも深刻なのは、流通経路において偽薬が混ざることである。
聞くところによると、世界に流通している医薬品の半分は偽薬という。さらにインターネットで取引されている某薬剤に至っては、90%が偽薬と言われている。
医薬品の流通におけるトレーサビリティが求められている。
したがって、製薬企業が苦情や有害事象報告を受けた場合には、まず自社薬であるかどうかをまず検証しなければならない。
方法はこうである。個装毎にキーコード(Microsoftのライセンスキーのようなもの)を印字しておき、苦情や有害事象報告があった際には、そのキーコードを聞き取り、システムに入力するのである。その際に、システムが自社薬であるか偽薬であるかを判別することになる。
回収の手順とその適格性検証
また、ディストリビューションでは、回収に関する手順の確立も重要である。
多くの製薬企業では、回収に関する手順書を作成済であると思われる。
しかしである。はたして、その回収に関する手順書の適格性を検証したであろうか。
PIC/S GMPでは、作成した手順書は、必ずその適格性を検証する必要がある。つまり、実際にテストを実施することが求められている。
かつてこんな恐ろしいエピソードがあった。
2004年12月3日、パリ郊外のシャルル・ドゴール空港で、警察官が爆発物探知犬の訓練のために乗客の荷物に爆発物をしのばせたところ、誤って荷物が国際線の旅客機に積み込まれてしまった。仏当局の通報を受けて米国などで到着便の大捜索が行われたという。
結果的には爆発物は発見されなかった。
欧州では、このように実際の状況をシミュレーションして訓練が行われる。恐らくそういう文化なのであろう。
本邦においても、PIC/S査察が行われた際には、絵に描いた餅であるSOPは認められず、適確性を確認した記録が求められることになるであろう。
回収だけではなく、災害対策(DRP)や業務継続計画(BCP)も同様に、実際にテストを実施し、その適格性を検証しておかなければならない。
PIC/S GMPと自己点検
GMP症例やコンピュータ化システム適正管理ガイドラインには、自己点検という要求事項がある。
ここで「自己」とは「私」ではないことに注意が必要である。
自己点検は、PIC/S GMPでは、Self Inspectionと表記されている。
これまでの製薬業界は、とりわけ本邦においては、当局の指摘に従って改善していれば、許可が与えられた。
しかしながら、そのような状況では、明らかに当局の指摘を行う能力や、時間に依存することになる。
当局に指摘された事項のみを改善するというのでは、患者やユーザの安全性を担保することは困難である。
従って、当局の査察(Authority Inspection)ではなく、自社の監査(Self Inspection)により、リスクを受容可能まで低減することが求められるのである。
自己点検は、内部監査、外部監査、定期レビュ、マネージメントレビュなどからなる。
GMP調査における査察官の関心は、問題をどう解決したかではなく、残存するリスクがないかどうかである。
自己点検により、自らリスクを発見、低減させることができず、当局の査察官がに指摘されるようでは困る。
PIC/S査察における監査報告書の調査
これまで一般的に日本や米国の査察では、監査報告書は調査されなかった。
これは監査の独立性を担保するためである。もし規制当局が監査報告書を調査するとなるとどうであろうか。恐らく多くの企業では、発見したエラーを正直に記載しなくなるのではないだろうか。
ところが、欧州の考え方は180度異なる。欧州での査察では、監査報告書を積極的に調査するのである。
その際に、製薬企業が自ら発見し改善措置を実施したエラーやリスクについては、規制当局は指摘を行うことはない。したがって、製薬企業は積極的にエラーやリスクを発見し、改善措置を実施することになる。
今後、PIC/S査察が実施されるようになれば、監査報告書(内部監査、供給者監査、サプラーヤオーディット)を提示する機会が増えることと思われる。
ところがである。果たして監査報告書が査察に耐えうる品質で作成されているかが問題となる。
製薬企業において、監査報告書が最も監査されていない文書であることは間違いない。他者とりわけ規制当局に提示できる品質で監査報告書が執筆できるであろうか。
PIC/S査察官は、いわばプロの集団である。百戦錬磨、多くの製薬企業を査察し、またPIC/S加盟国同士での事例交換も活発に行われている。
そのプロともいえるPIC/S査察官と同等の指摘(リスクの発見)ができるかどうかということが今後問われることになる。
PIC/S査察対応には、優れた監査担当者の育成または採用が不可欠であるといえる。
CAPAの重要性
FDA査察とCAPA
GMP調査における査察官の関心は、問題をどう解決したかではなく、残存するリスクがないかどうかである。
一般にエラーを発見した場合、速やかに修正が行われる。しかしながら、「問題を解決」しただけではダメなのである。問題の根本的原因を突き止め、再発防止を図ることが最も重要である。
そこでCAPAが重要となる。
CAPAとは Corrective and Preventive Action(是正措置・予防措置)のことであり、FDA査察で指摘の一番多いものの一つである。つまりFDA査察準備の最大のポイントである。
CAPAは不具合の調査、理解、修正に焦点を置く一方で、その再発の予防を試みるcGMP規則の概念としてよく知られている。
このCAPAを導入することにより医薬品製造企業における不適合の発生率を確実に減少させることが出来る。
CAPAの考え方は医薬品業界の査察のために米国FDAが開発し、その手順は品質に関する規制が適用となる品質システムの中で最も重要なものとなった。
FDAは「CAPAを理解していない会社が多い、特に海外の会社はこれを全く理解していない。」 「最大の問題は、CAPAの手順が適切でなく効果もないことだ。」などと見解を述べている。
品質システムにおいては、マネジメント・レビュの結果は通常記録される。計画されたアクションは、実効的な修正、予防アクションおよび変更管理プロシージャを利用することによって実施される。
CAPAは、マネージメントレビュなどを通して、経営者に伝えられ、QMSの改訂へとつながる。
CAPAにおけるよくある課題
筆者が経験する中では、CAPAの情報管理が、手作業ベース(Excel、Word)で行われ、関連する資料と共に紙ファイルで保存されているケースが多い。
これでは蓄積したデータの利用効率が悪く、CAPAの品質向上への効果が十分に得られない。
またCAPAと称して、多くの場合は顧客苦情のみを対象にしているケースが目立つと感じている。
FDAの要求事項であるCAPAでは、顧客苦情のみならず生産工程等での不適合にも適切な対応が求められている。つまり、内部監査、外部監査、QC、定期レビュ、マネージメントレビュなど様々な場面においてCAPAが導入されなければならないのである。
また、CAPAシステムを自社開発しているケースも多くみられるが、FDAの規制要件21 CFR Part 11に適合していない場合がほとんどである。


