プロセスバリデーションとは? ㈱イーコンプライアンス
プロセスバリデーションとは?
FDAは、1987年に発行した「Guidelines on General Principles of Process Validation」の中で、医薬におけるバリデーションとは「あらかじめ定めた仕様や品質にあった製品を継続的に生産するプロセスに対して、高度の保証を与え、文書化された証拠を確立するものである。」と定義している。
医薬品の場合、医療機器と違って製品の品質試験はその大半が破壊検査しかできない。
従って必然的に抜き取り(サンプリング)検査となってしまう、しかしながらサンプリング検査には問題がある。
例えば、1ロット10,000錠を製造したとしよう、そこから50錠をサンプリングして破壊検査を行い、異物が混入していないことを確認したとしても、残り9,950錠のどこかに異物が混入しているかも知れないのである。
これがサンプリング検査の問題である。
そこで、規制当局は医薬品の製造においてあらかじめバリデーションしておくことを求めている。
下図において、「不安定なプロセス」は、ロット毎に品質が変動している。
また最終的に変動幅が増大している。
もしこれがあんパンやジャムパンなら構わないかもしれない。
しかしながら医薬品においては、その種類によっては患者に多大な健康被害をもたらすことになるであろう。
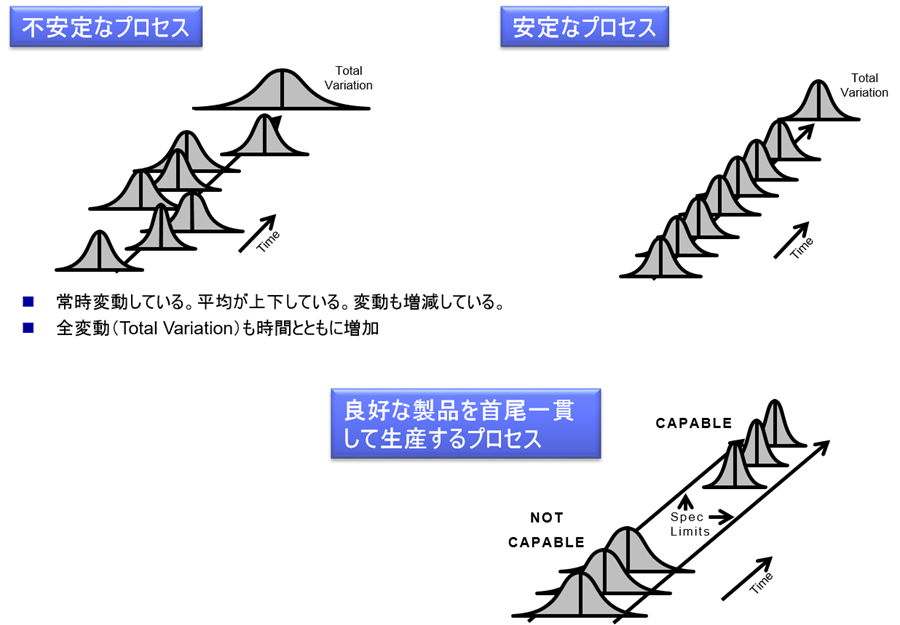
そこで、毎ロットの品質を安定させる必要がある。(安定なプロセス)
しかしながら、医薬品のようなプロセス産業(装置産業)においては、季節変動などの要因のため、その品質が変動する。
- 温度/湿度/機器の外装・結露
- 電力供給上の変動/振動/光
- 環境汚染/プロセス水の純度/人的要因
- 原料の銘柄違い(原料ロット切り替わりによる原液粘度変動―製品の除去率性能に影響)
つまりプロセス産業はディスクリート産業と違って、季節によっては1tの原材料を投入しても、製造された製品が1.1tになったり0.9tになったりするのである。
構造設備の経年劣化によっても、製品の品質が劣化することもあり得る。
またプロセスを安定させるのみではなく、「良好な製品を首尾一貫して生産するプロセス」においては、ロット内のすべての製品が規格内(Spec Limits内)に納まらなければならない。
医薬品の場合、サンプリング検査を実施し、規格外(OOS:Out of Specification)が発見されると、場合によってはロットごと廃棄しなければならない。
プロセスが恒常的に規格に合格した製品を通常の操作条件において生産できることがプロセスバリデーションでの目的である。
つまりプロセスの再現(繰り返し)性、長期のプロセス安定性を保証するのである。
プロセスバリデーションにおいては、確立した実生産の条件で製品を製造する。
同時に様々なアクションレベル、それを含んだ標準操作手順書(SOP)の内容を確認し、チャレンジテストの繰り返しでよりプロセスの保証を高める必要がある。
1987年当時、FDAはプロセスバリデーションを最低3ロット実施するよう要求していた。
なぜ3ロットであったかというと、2ロットでは直進性が分からないためである。
3ロット製造してみてはじめて直進性が判別できるのである。
しかしながら最新の規制要件では、もはや3ロットでは認められなくなっている。
バリデーションとベリフィケーションの違いは、バリデーションは「定義された品質・仕様の通り製品が製造できることをあらかじめ保証すること」に対して、ベリフィケーションは「定義された品質・仕様の通り製品が製造できたことを確認すること」である。
つまりバリデーションは未来形であり、ベリフィケーションは過去形である。
医薬品の製造では、従来の「バリデーション」+「サンプリング検査」から、QbD(Quality by Design)による連続的ベリフィケーションが要求される時代となった。
しかしながらバリデーションを連続的ベリフィケーションに置き換えるためには、製剤開発時にQbDによる開発を実施し、新薬製造承認時にQbDによる製造の承認を受けておかなければならない。
FDAは、PAT(Process Analytical Technology)と呼んでいる。
用語集

