
1. はじめに
2008年10月29日に開催された日薬連主催の「第28回医薬品GQP・GMP研究会」では、「コンピュータ化システムバリデーションのガイドライン」についての概要が発表された。
これは平成4年に発出され、平成17年3月30日に取り下げられた「コンピュータ使用医薬品等製造所適正管理ガイドライン(薬監第11号)」を改定するものである。
厚生労働省は、平成17年3月30日に本ガイドラインを取下げた。取下げの意図は不明である。
この取下げにおいて、輸出等において混乱をきたした。例えば、台湾行政院衛生署は、1999年5月1日に「薬品優良製造規範」を公告し、台湾市場へ医薬品を供給する台湾内外の製薬メーカに対しバリデーションの資料を段階的に提出することを義務付けた。そして2005年12月10日までに輸入許可を取得している全医薬品について、コンピュータバリデーションデータの提出を要請した。
そこで、厚生労働省医薬食品局監視指導・麻薬対策課は、平成18年10月13日付の事務連絡において「GMP/QMS事例集(2006年版)について」の117頁GMP 20-12 (コンピュータの利用等)において、新たなガイドラインが発出されるまでの間は、従来どおり、本ガイドラインを参考とすることとし、一部改定の上、現在でも有効とした。
その上で2007年6月より、厚生労働省、医薬品医療機器総合機構(PMDA)、日本製薬団体連合会 品質委員会、製剤機械技術研究会が共同で、本ガイドラインについての見直し作業を進めている。
2008年2月には、見直しにあたり、各企業の現状を踏まえたものとするため、日薬連及び日本医薬品原薬工業会傘下会員社にアンケート調査を実施した。
また改定案に対するコメント収集及び評価を行い、改定案の最終化と規制当局へ提案が行われた模様である。
今後は、厚労省版「コンピュータ化システムバリデーションガイドライン」が施行され、それにともなって査察マニュアルも見直しされるものと推察される。
今回は2008年10月29日 日薬連主催「第28回医薬品GQP・GMP研究会」配布資料をもとに、近い将来に発行されると予想される厚労省版「コンピュータ化システムバリデーションガイドライン」を考察してみたい。
2. 日本におけるコンピュータ化関連指針
日本におけるコンピュータ化関連指針としては、厚生省(当時)から平成4年2月21日に出された薬監第11号監視指導課長通知「コンピュータ使用医薬品等製造所適正管理ガイドライン」と、平成5年1月11日に出された薬監第3号監視指導課長通知「コンピュータ使用医薬品等製造所査察マニュアル」があげられる。
この2つの通知を受けて、平成5年に薬事法が改正され、医薬品GMPの改正が行われ、バリデーションを実施することが医薬品製造の許可要件となった。(図1参照)
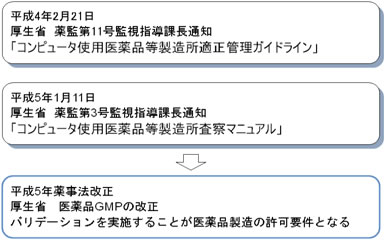
図1 日本におけるコンピュータ化関連指針
コンピュータ使用医薬品等製造所適正管理ガイドラインは、厚生省がソフトウェアベンダーに作成を依頼した経緯があり、実用的で、読みやすく分かりやすいものになっている。
しかしながら、本ガイドラインはソフトウェアを開発する側の論理で作成されており、検証する側の論理、すなわちバリデーションの概念が薄いものとなっている。(図2参照)
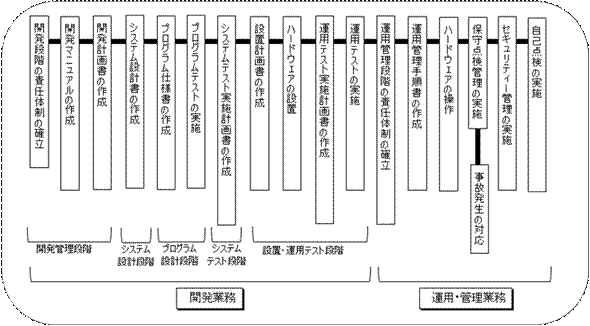
図2 コンピュータ使用医薬品等製造所適正管理ガイドライン
3. コンピュータ使用医薬品等製造所適正管理ガイドラインの取下げと復活
厚生労働省は、平成17年3月30日に突如本ガイドラインを取下げた。取下げの意図は不明である。
この取下げにおいて、輸出等において混乱をきたした。
例えば、台湾行政院衛生署は、1999年5月1日に「薬品優良製造規範」を公告し、台湾市場へ医薬品を供給する台湾内外の製薬メーカに対しバリデーションの資料を段階的に提出することを義務付けた。
そして2005年12月10日までに輸入許可を取得している全医薬品について、コンピュータバリデーションデータの提出を要請した。
そこで、厚生労働省医薬食品局監視指導・麻薬対策課は、平成18年10月13日付の事務連絡において「GMP/QMS事例集(2006年版)について」の117頁GMP 20-12 (コンピュータの利用等)において、新たなガイドラインが発出されるまでの間は、従来どおり、本ガイドラインを参考とすることとし、一部改定の上、現在でも有効とした。(図3参照)

図3 適正管理ガイドラインの「復活」
4. ガイドラインの見直し
輸出等で混乱をきたしているため、本ガイドラインの見直しは迅速に行われなければならない。
2007年6月より、厚生労働省、医薬品医療機器総合機構(PMDA)、日本製薬団体連合会 品質委員会、製剤機械技術研究会が共同で、本ガイドラインについての見直し作業を進めている。
2008年10月29日に開催された、日薬連主催の「第28回医薬品GQP・GMP研究会」では、「コンピュータ化システムバリデーションのガイドライン」についての概要が発表された。
5. 新ガイドライン検討の過程と今後の見通し
新ガイドラインの作成にあたって、以下のようなステップで見直し作業が行われた。
1) 基本的な方針の検討
2) 適正管理ガイドラインとGAMP 4比較検討
GAMP 4は、これまで事実上のグローバルスタンダードとして、世界中の製薬企業で使用されてきた。日本におけるCSVガイドラインが、グローバルスタンダードと乖離したものになった場合、日本の製薬企業にとっては、いわゆるダブルスタンダードという問題が起こってしまうことになる。したがって、GAMPに準拠したものにすることは極めて重要である。GAMPは、2008年2月に7年ぶりに改定版が発表され、GAMP 5が発行された。GAMP 5では、大幅な改定が行われた個所もあり、もし厚労省版「コンピュータ化システムバリデーションガイドライン」が、GAMP 4に沿って作成されているとすれば、問題が残ることになる。
3) CSV取り組み状況と課題の把握(日薬連、原薬工加盟企業へのアンケート調査の実施)
2008年2月には、ガイドライン見直しにあたり、各企業の現状を踏まえたものとするため、日薬連及び日本医薬品原薬工業会傘下会員社にアンケート調査を実施した。
日本の製薬企業の特徴として、中小・零細企業を含めると非常に多くの製薬企業が存在する。
そのすべての企業に対して、グローバルスタンダードの基準でコンピュータ化システムの品質保証を行うよう要求することは、困難を伴う。
本アンケート結果は、一般雑誌に詳細なグラフとともに発表された。
4) ソフトウェアカテゴリ分類の検討(製薬企業における分類の調査)
GAMPでは、ソフトウェアをカテゴリ分けしている。
GAMP 5では、GAMP 4までにはあった、ファームウェア(カテゴリ2)が削除された。
ファームウェアというのは、ICチップに焼付けたコードのことである。
その特徴は、一旦ICチップに焼きつけてしまうと、当該サプライヤでも変更することができない。
GAMP 5の改定では、この分類方法に異議が出された。何故ならば、ファームウェアといってもICチップに焼付けるまでは、ソフトウェアである。つまりバグ等の不具合もあれば、変更管理も行われるのである。
GAMP 5では、最終形がファームウェアのように変更できないかどうかという、形態によって分類はしなくなった。
5) 適正管理ガイドラインの改定版検討
改定版は、GAMP 4で使用されてきた用語をもとにしているようである。
例えば、IQ、OQ、PQ、DQなどである。
これらの用語は、GAMP 5では使用しないこととなった。
6) 改定案に対するコメント収集及び評価
日薬連加盟の各製薬会社に、改定案に対するレビュとコメントを求めた。
しかしながら、各社に対してコメントに対する回答等は、行われなかった模様である。
7) 改定案の最終化と当局へ提案
改定案はすでに当局に提出されたものと推察する。
また当局が発行する際には、パブリックコメントを募集するよう要請した模様である。
8) 解説書の作成(査察マニュアルの見直し)
現在、当局側では査察マニュアルの作成を行っているものと推察される。
6. ガイドライン見直しのポイント
医薬品GQP・GMP研究会での発表によると、ガイドライン見直しのポイントは4つあるという。(図4参照)

図4 ガイドライン見直しのポイント
6.1 CSVにかかる労力やコストの削減
GAMPと整合させ、ソフトウェアをカテゴリ分類することにより合理的・経済的なCSVとしなければならない。
ソフトウェアの変更度合いに応じて、カテゴリを分類する。変更が多いほど不具合等のリスクが増す。すなわちカテゴリ分類は、リスク分類である。
リスクの大きいソフトウェアは、従来通りのCSV実施が求められるが、リスクが低いものに関しては、CSV実施の程度を抑えることができるのである。
これにより、効率的、効果的で、コスト効率の良いコンピュータ化システムの品質保証が行えることとなる。
6.2 グローバル化への対応(欧米とのハーモナイズ)
日本の製薬企業にとって、色々な規制要件において、いわゆるダブルスタンダードという問題が多く発生している。
例えば、これまでのコンピュータ使用医薬品等製造所適正管理ガイドラインとGAMP 4の関係や、21 CFR Part 11と厚労省ER/ES指針の関係等である。
新しいガイドラインにおいては、GAMP 5と整合し、ダブルスタンダードを起こさないよう配慮することは、必須の課題である。
6.3 CSV規定とリスクアセスメント・サプライヤ評価
新ガイドラインのもとでは、製薬各社は、CSV実施のための規定(ポリシー)やSOPを作成することが必須となる。
また世界のCSV規制の動向として、リスクアセスメントを実施することは必須である。
さらにCSVの実施においては、サプライヤを最大限活用し、コストや労力を下げ、品質を向上させることも重要である。
これらリスクアセスメントや、サプライヤアセスメントに関する文書(SOP)も、作成しなければならない。
特にサプライヤを利用するには、前もってサプライヤ監査を実施し、当該サプライヤのQMSを監査しておかなければならない。
6.4 適用範囲の明確化と取り組みやすいCSV
これまでPLC(Programmable Logic Controller)の取り扱いについては、曖昧であった。英国のようにPLCに関して厳しいCSVを求めている当局もあれば、そうでない国もあった。
このように不明確であったCSVの適用範囲を、明確にすることは重要である。
また、これまで製薬業界におけるバリデーション作業は、大変難しい用語が使用されてきた。
例えば、据付時適格性検証(IQ)、稼動性能適格性検証(OQ)、稼動時適格性検証(PQ)などである。
通常、ソフトウェア業界では、検証(Qualification)という用語は使用しない。一般にはテストと呼ぶ。
検証という言葉は、完全な一致性の確認と言ったニュアンスに聞こえ、CSVを面倒くさく、複雑で、難しいものと感じさせる要因となっている。
判りやすい表現と、説明書の作成は必須事項である。
7. カテゴリ案
新ガイドライン(案)では、GAMPと同様、ソフトウェアをカテゴリ分けしている。(図5参照)
カテゴリはすなわちソフトウェアのリスクの分類である。カテゴリの数字が大きくなるほど、ユーザによる変更の度合いが大きく、ソフトウェアに欠陥が含まれる可能性が増大する。
カテゴリ |
内容 |
一般例 |
||
0. |
機能の固定された汎用品 |
商業ベースで販売されている汎用機器 |
電話機、電卓、時計、ストップウォッチ、タイマー等の機器 |
|
1. |
基盤ソフト |
階層型ソフトウェア(カテゴリ3以降のアプリケーションが構築される基盤となるもの) |
OS、データベースエンジン、ミドルウェア、プログラム言語、統計管理パッケージ、ネットワーク監視ツール、スケジュール管理ツール |
|
3. |
構成していないソフトウェア |
商業ベースで販売されている既製のソフトウェアで、ソフト自体は業務工程に合わせて構成していないもの。(実行時のパラメータ入力、パラメータの記録は本カテゴリに含まれる) |
市販のパッケージソフトウェア |
|
4. |
構成したソフトウェア |
ユーザの業務プロセスに合わせて構成したソフトウェア。 |
LIMS、SCADA、MRPII、MES |
データ収集システム、ERP、DCS |
EDMS(文書管理システム)、倉庫管理システム、表計算ソフト上の演算テンプレート等 |
||||
5. |
カスタムソフトウェア |
業務プロセスに合わせて設計され、プログラムされたソフトウェア |
ITアプリケーション、プロセスアプリケーション、カスタムラダーロジック(PLC)、カスタムファームウェア、表計算等のマクロプログラム |
|
図5 カテゴリ案
71 カテゴリ0 機能の固定された汎用品
カテゴリ0は、GAMPにはない。電卓やストップウォッチなどのCPUを搭載しているだけのものや、MS-Office製品等をインストールしているのみのPCが相当する。
7.2 カテゴリ1 基盤ソフト
カテゴリ1は、基盤ソフトであり、GAMP 5ではInfrastructure Softwareと呼んでいる。GAMP 4ではOperating Systemであった。
これにはOS、OracleやSQL Serverのようなデータベース、ミドルウェア、ネットワーク監視ソフトなどを含む。
7.3 カテゴリ2 使用しない
カテゴリ2は、GAMP 4ではFirmwareであったが、GAMP 5では使用しないこととなった。
これに合わせて、新ガイドライン(案)でもカテゴリ2を欠番としている。
ファームウェアというのは、ICチップに焼付けたコードのことである。その特徴は、当該サプライヤでも変更することができない。しかしながら、ファームウェアといってもICチップに焼付けるまでは、ソフトウェアである。つまりバグ等の不具合が存在し得るのである。
GAMP 5では、最終形がファームウェアのように変更できないかどうかという形態によって分類はしなくなった。
ファームウェアは、後述するカテゴリ3から5のいずれかのソフトウェアに組み込まれて使用されているか、一緒にシステムを構成しているはずで、ファームウェア単独でのCSVは実施しないことになった。
7.4 カテゴリ3 構成していないソフトウェア
カテゴリ3は、構成していないソフトウェアであり、GAMP 5ではNon-configured Softwareである。GAMP 4では、Non Configurable Software(構成設定不可なソフトウェア)であった。
別名COTS(Commercial Off The Shelf)として知られている。Shelfは棚のことであるが、Off The Shelfとは、棚から降ろしたらすぐに使用することができるという意味である。つまりユーザ設定や権限設定などは不要で、またワークフローの設定もしない。
これは設定変更不可であるソフトウェアや、設定変更が可能(Configurable Software)であっても、設定変更していない(工場出荷時のままの値で使用する場合)ものが含まれる。
GAMP 4では、設定変更しなくても、設定変更可能なソフトウェアである場合、カテゴリ4に分類していた。GAMP 5では、設定変更したかしなかったかでカテゴリを分けることになった。
カテゴリ3の例をあげると、電子天秤等、設置すればすぐに使用できる分析機器等である。またMS-Excelをそのまま使用する(ワークシートに数値等を入力する)場合が相当する。
7.5 カテゴリ4 構成したソフトウェア
カテゴリ4は、構成したソフトウェアであり、GAMP 5ではConfigured Softwareである。GAMP 4では、Configurable Software(構成設定可能なソフトウェア)であった。
これはビジネスプロセスに合わせて、パラメータなどを変更し、機能を変更しているものである。つまりカスタマイズしないで、ユーザの意図した仕様に適合させることができるものである。
昨今のソフトウェアのほとんどは、このカテゴリ4に相当する。
カテゴリ4の例としては、ドキュメント管理システムのように、ワークフローを設定し、ユーザ設定と権限設定によって、著者、レビューア、承認者等を区別し、プロセス通りの機能を実現するものがあげられる。
またMS-ExcelでTemplate等を使用し、縦と横の罫線を引いた表に入力した際に、合計や平均を自動的に計算させるような設定をした場合があげられる。これはMS-Excelの関数をコールしているのであって、カスタマイズやプログラミングを行っているわけではない。
7.6 カテゴリ5 カスタムソフトウェア
カテゴリ5は、カスタムソフトウェアであり、GAMP 4や5ではCustom Softwareである。
パッケージをカスタマイズした場合や、自社開発したソフトウェアが相当する。
またMS-Excelで、マクロやVBAと呼ばれるBasicプログラムを作成した場合が相当する。
8. カテゴリ毎の対応
カテゴリ |
一般的対応 |
|
0. |
機能の固定された汎用品 |
CSV対象外 |
1. |
基盤ソフト |
バージョン番号の記録 |
3. |
構成していないソフトウェア |
リスクに基づき簡略化したCSVを実施する |
4. |
構成したソフトウェア |
リスクに基づきCSVシステムを一部簡略化してもよい |
5. |
カスタムソフトウェア |
ガイドラインに記載されたCSVプロセスの実施 |
図6 カテゴリ毎の一般的対応
8.1 カテゴリ0 機能の固定された汎用品
カテゴリ0は、CSVの対象とはならない。したがって、設置時にパッケージソフトウェアに関しては必要に応じてバージョン番号、PCの機種番号、製造番号の記録等を行うのみである。
8.2 カテゴリ1 基盤ソフト
カテゴリ1も、カテゴリ0と同様の扱いとなる。
一般にインフラストラクチャに関しては、CSVの対象とはならないが、Qualification(検証)の実施は求められる。
すなわち、サーバ、ルータ、ハブの検証などである。CSVと違う点は、テストデータを入力し、仮説検証型で検証するのではなく、それら装置が仕様の通り稼働することを確認することである。
ネットワークの設定等についても同様に、正しくアドレスがふられていること、フィルタリングが正しく設定されていることなどを検証する。また最近使用される機会が多くなった、ワイヤレス装置についても、目視では点検できないため、正しく当該装置と接続されていることを検証しておかなければならない。
8.3 カテゴリ3 構成していないソフトウェア
カテゴリ3は、ほぼCSVを簡略化できると考えてもよい。
特にGAMP 4におけるOQの実施はほとんど必要性がないと思われる。
例えば、Excelをそのまま使用したり、電子天秤を設置する場合などであるが、それぞれ機能をテストすることはあまり考えにくい。ただし、GAMP 5におけるPQの実施は、リスクレベルに応じて実施するべきである。
すなわち、実際の業務で支障なく使用することができるかどうかを確認しておくのである。
電子天秤の場合、IQ(適切な場所に設置されていることを確認、バージョン・製造番号等の記録等)を実施したのち、旧来のものと同等性があるか、精度が十分か、キャリブレーションが問題なく行えるか、実際の業務で使用する際に問題となる点はないかというような、確認のみを行うことになるであろう。
供給者監査(サプライヤーオーディットを直訳していると思われる。日本ではベンダーオーディットと呼ぶ場合が多い。)は、リスクベースで実施することになる。
カテゴリ3において供給者監査を実施しなければならないリスクとしては、当該コンピュータ化システムに対して、最初のユーザとなる場合などが考えられる。
ExcelやSASなどの汎用的なソフトウェアに関しては、大量に使用されていることもあり、リスクは低いが、パッケージ製品とは言え、新規性のあるものに関しては、その導入はリスクを伴う。
しかしながら、一般的には、カテゴリ3の供給者に対する監査は行わないと考える方が一般的と思われる。
8.4 カテゴリ4 構成したソフトウェア
リスクに基づき、CSVを一部簡略化して実施することとなる。
具体的には、自社でパラメータやコンフィグレーションを設定し、機能を変更したもののみを対象として、GAMP 4におけるOQを実施することになる。
パッケージ製品の場合、自社で変更していない機能は、当該サプライヤが出荷前にテストを実施し、その品質を保証しているため、テストを二重化して実施する必要性はない。
供給者監査についても、リスクに応じて実施することとなる。対応はカテゴリ3の場合と同様である。
GAMP 4におけるPQの実施は必須である。コンフィグレーション後の機能(すなわち当該業務に適合させるようパラメータ等を変更した機能)が、正しく当該業務に対して支障なく利用できることを確認することとなる。
CSVに関する文書・記録類は、製薬企業で保存しておかなければならない。ただし、設計仕様書等、システム構築に関する文書・記録類は、当該サプライヤが管理してもよい。必要に応じてそれら仕様書等を、供給者監査で調査しておくこと。
運用のための手順書の作成は、必須である。また手順書を遵守して運用することも重要であることは言うまでもない。
8.5 カテゴリ5 カスタムソフトウェア
カテゴリ5の場合、自社のCSV SOPに従った、フルのCSV実施が求められる。
言い換えれば、カテゴリ3や4は、リスクベースにより、SOPに記載されているアクティビティや作成成果物を一部割愛できるということである。
カテゴリ5の場合、供給者監査は必須である。契約前には終えておく必要がある。また監査に合格できていないにもかかわらず、契約を締結してはならない。
GAMP 4におけるDQ(Design Qualification:設計時適格性確認)の実施も必須である。DQとは、設計がユーザの要求を満たしていることを、製造に前もって検証しておくことである。
DQの実施にあたっては、当該システムに関する深い知識と経験が必要となるため、当該サプライヤが実施することとなる。
製薬企業側では、そのDQが適切に行われたかどうかを確認しておく必要がある。特にトレーサビリティマトリックスの作成が行われており、ユーザ要求仕様書で要求された機能が、もれなく設計仕様書に反映されていることを確認しておくこと。
また当該サプライヤは、ソースコードを作成した際に、ソースコードレビュを行わなければならない。
ソフトウェアは、テストデータの入力によるテスト(一般にブラックボックステストと呼ばれる)だけでは、すべての条件分岐をテストすることができないため、デバッギングツール等を用いた目視によるソースコードレビュ(一般にホワイトボックステストと呼ばれる)が必要である。
その際のレビュ担当者は、当該プログラマではなく、別の者が担当しなければならない。なぜならば当該プログラマは、当該ソースコードに対する“思い込み”があるからである。
表中に第三者と記載があるが、これは当該サプライヤ内の別人物と解釈しても良いと思われる。
9. 新CSVガイドライン目次案
新CSVガイドラインの目次案は、図7のようになっている。
注目すべきところは、3章の「 CSV規定の作成」であり、厚労省版CSV指針が発出された後は、CSVに関するポリシーやSOPを作成しておくことが必須となる。
4章の「開発業務」は、コンピュータシステムを開発する者が理解しなければならない。おもにサプライヤがその任にあたるものと思われる。
5章の「検証業務」は、バリデーションのことであり、システム開発・導入に関わるすべての者が理解しておく必要がある。
GAMP 5(英語版)では、Qualificationという用語を廃し、Verificationに改めた。しかしながら本ガイドライン案では、検証(Qualification)という言葉遣いや、DQ、IQ、OQ、PQといったGAMP 4での用語が使用されている。このまま発行されると、GAMP 5に従ってSOPを作成した企業では、齟齬が生じてしまう懸念がある。
厚労省が最終版を発出する際には、GAMP 5に日本語版と用語や翻訳の整合をとっていることを期待する。
0. 制定の経緯 |
5. 検証業務 |
図7 CSVガイドライン 目次案
10. システムのライフサイクルとCSV
一般にコンピュータ化システムの開発・導入は、各社のQMS(Quality Management System)に従い、システムライフサイクル(SLC)に沿って実施される。(図8参照)
CSV活動は、SLCの各時点で実施されなければならない。CSVは、けっして後付けで実施してはならない。
SLCにおけるCSV実施は、仕様と確認のアプローチ(Specification and Verification)をとる。つまり「仕様」(Specification)は必ず「確認」(Verification)されなければならないのである。
ここで確認とは、パラメータやコンフィグレーションの読合せ(仕様書と実際の画面の確認など)である場合と、多くの場合はソフトウェアのテストを意味する。
GQP・GMP研究会で紹介された図は、GAMPにおけるV-Modelに相当するものであると理解する。
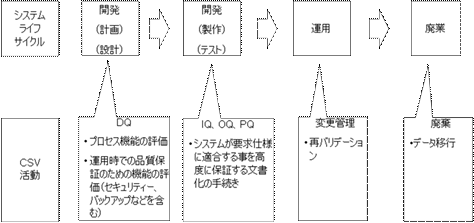
図8 システムのライフサイクルとCSV
また開発業務において、サプライヤを最大限活用することは、効率面、費用面、品質面において有効である。
プログラムやシステムのテストなどは、製薬企業が実施するのではなく、サプライヤに委ね、それらの記録もサプライヤが作成することになる。「もちはもち屋」なのである。
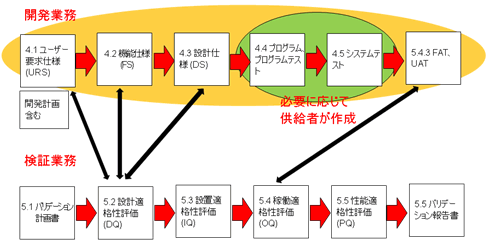
図9 CSVの流れ
11. 厚労省版CSV指針対応の課題
厚労省版CSV指針が発出された際に、製薬企業で対応すべき必須項目が3つ存在する。(図10参照)
◆ CSV取組みに対する社内体制の確立 |
図10 CSVの今後の課題
11.1 CSV取組みに対する社内体制の確立
製薬企業において、コンピュータ化システムの導入や運用を行う際に、CSVの取り組みに対する社内体制の確立が必要である。(図11参照)
社内体制は、平成17年に発出された厚労省ER/ES指針にも対応するものであることが望まれる。
「責任者」は、企業のトップまたは経営者であり、社内の品質保証体制全般に最終責任を負う。後述するコンピュータ化システムの品質保証ポリシー(CSV Policy)を作成し、全従業員に対し、周知徹底しなければならない。このことを一般に内部統制(Corporate Governance)と呼ぶ。
システムの信頼性保証に関するステアリングコミッティーとして、「CSV委員会」を設立する必要がある。
CSV委員会では、後述する企業内に存在するコンピュータ化システムのインベントリーを管理・メンテナンスする必要がある。また規制要件のモニタリング、社内のCSV実施状況の把握などにも責任を持つ。
必要に応じて経営者への報告を行う。
管理者は、実際にCSVの実施に関して、指揮を行う者である。CSVの実行責任を持つ。
スタッフ部門として、CSV SOP作成部門、教育部門があり、また日々のコンピュータ化システムのメンテナンス、バックアップ等を実施するIT部門が存在する。
コンピュータ化システムの品質保証を行う者としてCVQA(Computer Validation Quality Assurance)も必須である。
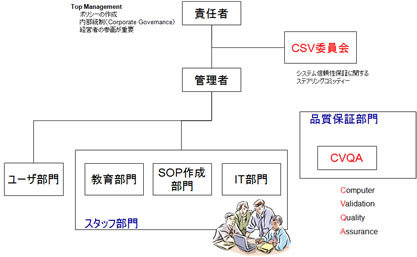
図11 CSV取組みに対する社内体制の確立
11.2 CSV取組み社内規定(方針、基準書、各種SOP)の作成
コンピュータ化システムの品質保証を行うため、QMSを構築しなければならない。
QMSを構築するということは、コンピュータ化システムに関する品質保証のための文書体系を確立するということである。
一般にQMSは、ポリシー(Policy)、役割と責任(R&R:Roles and Responsibility)、規則(Rule)、標準業務手順書(SOPs)、テンプレート(WPD)などから成る。(図12参照)
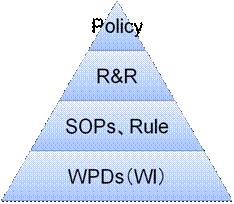
図12 一般的なQMSの階層構造
一般に、上位文書の方が概念的に記載し、下位文書の方が具体的に記載される。したがって、上位ほど改訂頻度が少なく、また上位レベル(例:社長や役員)の承認が行われ、下位ほど改訂頻度が多く、下位レベル(例:部門長)の承認で済ませることになる。
1)ポリシー(Policy)
ポリシーは、法や規制要件を遵守するための会社としての方針とコミットメントを記述する。したがって、ポリシーは全社で1つのものであり、その承認者は社長(または経営者)である。
ポリシーにはWhyを書くことになる。
ポリシー以下の文書は、ポリシーに記載された方針に相違することなく作成されなければならない。いわばポリシーは憲法的なものであるといえる。
一度制定されたポリシーは、法や規制要件が改定されることがない限り、変更するべきではない。ポリシーがしばしば変更されるとすれば非常に奇怪である。
ちなみに外国の会社による監査や、当局の査察では、このポリシーの提示と説明を求められることが多い。
経営者は常に自社の従業員に対して、このポリシーを徹底するよう周知しなければならない。このことを内部統制(Corporate Governance)と呼ぶ。
2)役割と責任(R&R)
役割と責任(R&R:Roles & Responsibilities)には、ポリシーによって会社がコミットした遵守事項を実行するために、権限委譲された組織や役割とそれらの責任を記述する。R&Rという権限委譲文書がなければ、全ての実施の責任は社長になってしまうのである。
つまりR&RにはWhoを書くことになる。ちなみに役割とは、バリデーションマネージャ、プロジェクトオーナーなどのような名称を指す。しばしば「役割」という用語と「責任」という用語が混同して用いられていることがあるので注意したい。
3)規則(Rule)
Ruleとは、規則のことである。例えば、「パスワードは6文字以上設定すること」や「パスワードは90日以内に変更すること」といった基準が相当する。
多くの場合、これらはSOPに含めがちであるが、そもそもSOPとは、ルールを守るための手順を記載するものである。ルールと手順は区別して作成することが望まれる。
4)標準業務手順書(SOPs)
SOPでは、R&Rで定義された役割が、その責任に応じて実施すべき手順を記載する。手順には、R&Rで定義した役割を主語とし、実施する時期、実施すべきタスクを明らかにする。
つまりSOPにはWho、When、Whatを書くことになる。
SOPには、フローチャートを記載すること。フローチャートが描けない場合は、おそらく記載事項が手順ではなく、ルールである可能性がある。
5)テンプレート(WPD)
テンプレート(WPD:Working Practice Document)では、CSVの記録を作成するための方法を詳細に記載する。製薬企業によっては、WI(Working Instruction)と呼ぶ場合もある。
WPDにはHowを書くこと。Howは経験をつむにつれ、改善され、進化する。したがって、WPDはしばしば改訂されることになる。
12.3 コンピュータシステムのインベントリー(台帳)作成
コンピュータシステムのインベントリーとは、社内に存在するコンピュータ化システムの台帳のことであり、一覧表である。(図13参照)
FDAにおける21 CFR Part 11やEMEAにおけるANNEX 11対応時には、このシステムインベントリーの作成管理が必須とされてきた。
システムインベントリーは、製薬企業内のどのプロセスやデータが電子化されているのかを一覧するものであり、CSV査察時において最初に説明を求められる事項である。
図13 システムインベントリー
システムインベントリーには、ハードウェアの名称と製造番号、ソフトウェアの名称とバージョンをはじめ、導入日、管理者名、どのような手順書を作成したかなどの記載も必要と思われる。
またCSVを実施したことを表す日付も記載しておくと良いだろう。
一般にシステムインベントリーを最初に作成する場合は、資産台帳などを参考にすれば効率的である。しかしながら、社内には個人が作成し使用しているMS-ExcelやMS-Accessなども多く存在することがあり、それらについても調査しておかなければならない。
またシステムインベントリーへの記載は、システムの導入後では遅いと思われる。企業は、コンピュータ化システムの導入を起案した段階で把握しておく必要があり、サプライヤー監査の実施、当該システムがPart11や厚労省ER/ES指針を満たすことの確認、CSVの実施等の状況をシステムライフサイクルの当初からシステムインベントリーへの記載をもって管理することが望まれる。
参考
- 「第28回医薬品GQP・GMP研究会」配布資料 日薬連 2008.10.29


