リスクベースドアプローチとは ㈱イーコンプライアンス
リスクベースドアプローチとは
リスクベースドアプローチとは
リスクベースドアプローチとは、「cGMPs for the 21st Century Initiative」(21世紀に向けたcGMPのイニシャティブ)の中で明らかにした、FDAの新しい医薬品業界監視の方針のことである。
1997年、FDAの行政改革を目的としてFDC法を改正する「FDA近代化法」がアメリカ議会を通過し、同年11月21日、クリントン大統領の署名によって施行された。
この改正法は医薬品と医療機器に関する規制の強化と緩和が中心となっていて、多くの規則の改正や制定、あるいはガイダンスの作成を要求し、これまでにない広範な改革を求めるものであった。
FDAは法律の発効後、3年以内に法律が要求する規則やガイダンスの仕事のほとんどを完了させた。
FDAは「cGMPs for the 21st Century Initiative」の包括的なプログラムの中で、品質マネジメントシステムとリスクベースアプローチに取り組むこととなった。
cGMPイニシアチブは、2002年に実質的に開始された。
2002年8月22日のFDA News Letterで「最初のゴールは、cGMP の要件を拡大し、追加の規制要件と当局のリソースをより重大な潜在的リスクを引き起こす生産の局面に適用することによって、公衆衛生に対する潜在的リスクに真正面から向き合うことである。」と宣言している。(図参照)

規制要件強化と患者負担のジレンマ
規制当局は、患者の安全性を担保するために規制要件を強化しなければならない。
しかしながら、規制要件を強化した場合、製薬企業にコンプライアンスコストの負担を強いることとなる。企業が支払ったコンプライアンスコストは薬価に転嫁され、結果的に患者負担になってしまう。
大げさに言えば「高額所得者しか救えない医療」になってしまうのである。

規制当局は、患者の安全性を担保するために規制を強化しなければならない反面、いたずらにコンプライアンスコストを高めてはならないのである。これはジレンマである
このジレンマを解いたのが「リスクベースドアプローチ」である。
リスクの高い医薬品の製造や品質試験にはそれなりのコンプライアンスコストをかけなければならないが、リスクが低い医薬品の場合、リスクに相応したコンプライアンスコストで良いというアプローチである。
規制当局は、リスクに応じて承認審査、査察を実施し、リスクに相応した指導を行うのである。
具体的には、製品が異なればリスクが異なる。
例えばビタミン剤や栄養剤などはリスクの低いだろう。一方、抗がん剤、向精神薬、ワクチン、血液製剤などはリスクが高い。
またプロセスが異なればリスクが異なる。
例えば、原材料の倉庫と最終製品の倉庫の温湿度管理では、最終製品の倉庫の方がリスクが高い。
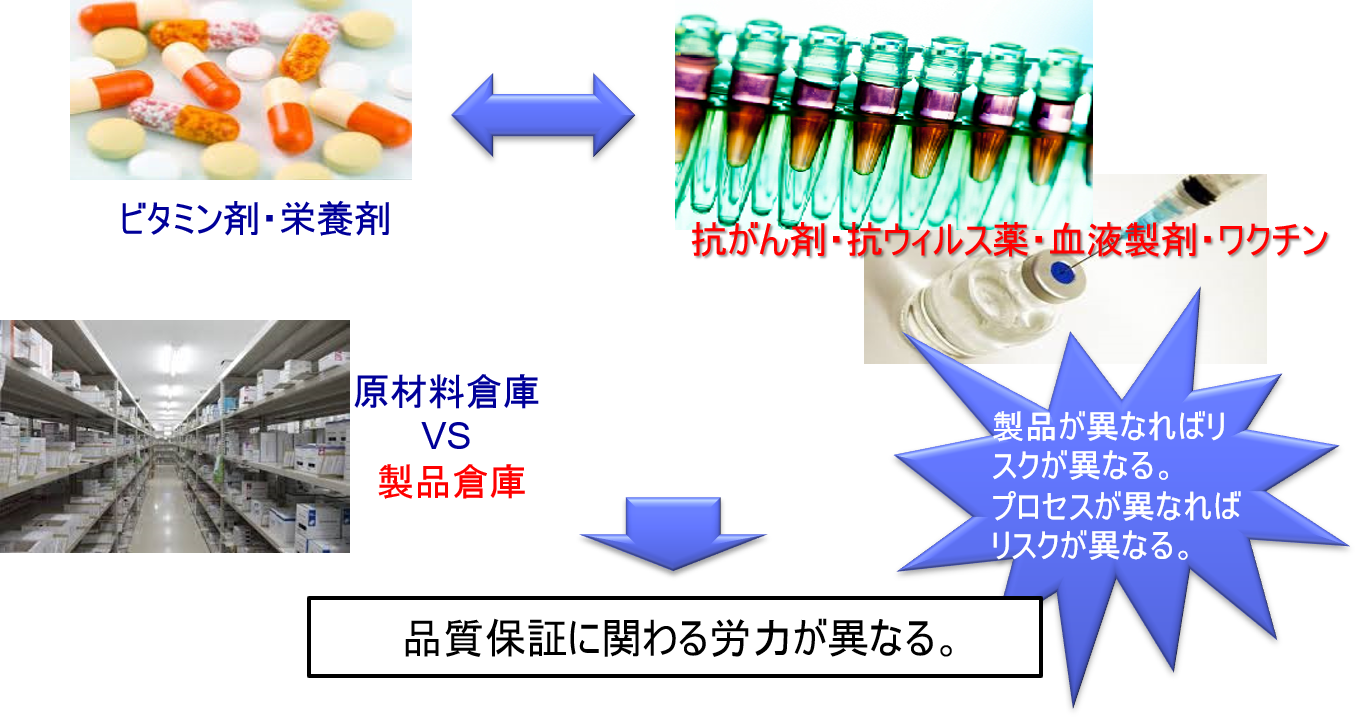
リスクベースドアプローチによって、規制当局の査察および製薬企業の品質保証にかかわるリソースの配分をよりリスクの高いプロセスに集中させることができるのである。
基準からリスクベースドアプローチへ
GMPは本邦において「医薬品等の製造管理および品質管理に関する基準」と呼ばれている。
すなわちGMPは基準である。しかしながら、どのような医薬品でも一定の基準に従って製造すれば良いというものではない。
昨今の規制要件のほとんどがリスクベースドアプローチになっている。
以前のように規制要件では「基準」を与えるのではなく、製薬各社が「リスク」に応じて自ら基準を定義しなければならない。
その際に重要なことは「リスクアセスメント」である。
自社が製造する製品のリスクを適正に評価し、リスクの大きさを調査しなければならない。
またすべてのプロセスの中で、どのプロセスがよりリスクが高いのかも調査しなければならない。
つまり規制当局は、正当化されたリスクアセスメントの文書化を要求しているのである。
Part11とリスクベースドアプローチ
2002年当時の統計によると、Part11を遵守するためにかかる費用(コンプライアンスコスト)は、全米の製薬企業で2,500億円とされた。
つまりPart11の発出によって、コンプライアンスコストを著しく増大させてしまった。
その為、FDAはコンプライアンスコストが他の規制要件に比べてはるかに高額となってしまうPart11に関しては、執行を裁量するということを2003年8月に宣言した。
現状ではPart11は、ヒト用の医薬品を製造する製造所のQCラボに限って査察が行われている。(医療機器、動物薬等ではPart11査察は実施されていない。)
その理由は、ラボにおける試験で、OOS(Out of Specification:規格外製品)になってしまった電子データ(分析結果や試験報告書)を改ざんして出荷した場合、患者の安全性に関わるからである。
医薬品においては、QCラボの試験結果および出荷判定のデータが最も患者への安全性に影響するため、リスクが高いのである。
リスクマネジメントに関する書籍
 |
【要点をわかりやすく学ぶ】 製薬・医療機器企業におけるリスクマネジメント |
|
≪ここがポイント≫ 医薬品・医療機器それぞれのリスクマネジメントを初心者にも解りやすく解説! ・リスクマネジメントの全体像の把握と個々のアセスメント手法の理解 ・リスクベースドアプローチとは?? ・FDAが求めるリスク管理と査察対応 ・医薬品・医療機器のリスクマネジメントの差異と各特徴・留意点 ・「リスク」と「ハザード」の違いと各特定方法、マネジメント手法 【発刊日】2015年8月28日 【著 者】株式会社イーコンプライアンス 代表取締役 村山 浩一 |
筆者が常日頃から思ってきたことは、医薬品(ICH-Q9)や医療機器(ISO-14971)に関するリスクマネジメントのセミナーや書籍が皆目ないということである。その理由は定かではないが、おそらくいずれも非常に難解であることと、網羅的に実践した経験者が圧倒的に少ないことに起因するのではないかと思われる。
本書では、医薬品と医療機器のリスクマネジメントを両方取り扱う。医薬品と医療機器では、リスクマネジメントに関する対応方法や対象が異なる。
しかしながらそのプロセスはほぼ同じである。
医薬品と医療機器で、どのようにリスクマネジメントの実施に差異があるかということにも言及した。本書では、難解なリスクマネジメントについて、できる限りわかりやすく執筆したつもりである。本書が、読者諸兄のリスクマネジメントへの理解を深める一助となり、より安全な医薬品・医療機器を世の中に出せることを願っている。
(はじめに 抜粋)
ご購入はこちら。
リスクマネジメントに関する規程・手順書・様式
 |
【ISO14971:2007対応】 リスクマネジメント規程・手順書・様式 |
| ISO14971:2007に沿った形のリスクマネジメント規程・手順書・様式です。 医療機器設計におけるリスク分析は、ISO-14971に従って実施されています。 リスクマネジメント実施のための手順や様式を整えておかなければなりません。 これから作成する医療機器企業やISO-14971認証審査を予定している企業、認証機関から改善指示を受けた企業向けに、サンプルをご用意いたしました。 MS-Word形式ですので、貴社でご自由に加筆・修正を行っていただけます。 |
≪様式一覧≫
※ご注文いただきますと、以下の様式を電子メールにて Wordファイル形式で納品いたします。
・MD-QMS-K5 リスクマネジメント規程
・MD-QMS-S501 リスクマネジメント手順書(ソフトウェアあり)
・MD-QMS-S501 リスクマネジメント手順書(ソフトウェアなし)
・MD-QMS-F501 ハザード項目検討票
・MD-QMS-F502 リスクマネジメントワークシート(ソフトウェアあり)
・MD-QMS-F502 リスクマネジメントワークシート(ソフトウェアなし)
・MD-QMS-F503 リスクマネジメント計画書テンプレート
・MD-QMS-F504 リスクマネジメント報告書テンプレート
ご購入はこちら。


