医療機器ソフトウェアの製造・販売をするためには ㈱イーコンプライアンス
医療機器ソフトウェアの製造・販売をするためには
2014年11月25日から、薬事法が一部改正される。
これにより、現在の「薬事法」という名称から、「医薬品、医療機器等の品質、有効性および安全性の確保等に関する法律」(医薬品医療機器等法)という名称に変更される。
医薬品医療機器等法のポイントはこちら。
日本国内で医療機器となるソフトウェアを設計・製造・販売するためには
改正法では、医療機器の「機械器具等」の範疇に、「ソフトウェア(プログラム)」が追加されといった大きな変更がなされた。
これまではソフトウェアはハードウェアに含められて、医療機器とみなされてきたが、改正法下では、プログラム単体でも医療機器となる可能性がある。
例えば、診断等に用いる単体プログラムについて、医療機器として製造販売の承認・認証等の対象となる。
したがって、改正法下で医療機器に該当するソフトウェアを開発する企業において、医療機器の製造販売業許可の取得や、製造業の登録が必要となった。
製造販売業
製造販売業については、高度管理医療機器、管理医療機器等の種類に応じて許可を取得する必要がある。(新法第23 条の2関係)
医療機器を市場に出す事業者(製造販売業者。輸入業者も含まれる。)は、医療機器の製造販売業許可を取得することになる。
医療機器を日本国内市場に出すにあたっては、医療機器の品質が保証され、ユーザーや患者、医療関係者等の安全が確保されるものでなければならない。
そのため、医薬品医療機器等法では、製造販売業許可の要件として、品質保証と安全管理の体制を整えることが求められている。
医療機器製造販売業許可は、「事業者」が取得する。
一法人にひとつの許可。(第1種医療機器製造販売業許可と第3種医療機器製造販売業許可を同時に持つことはない。)
複数の営業所がある場合は、総括製造販売責任者の常駐する事業所(本社等)がある都道府県知事に、許可を申請する。
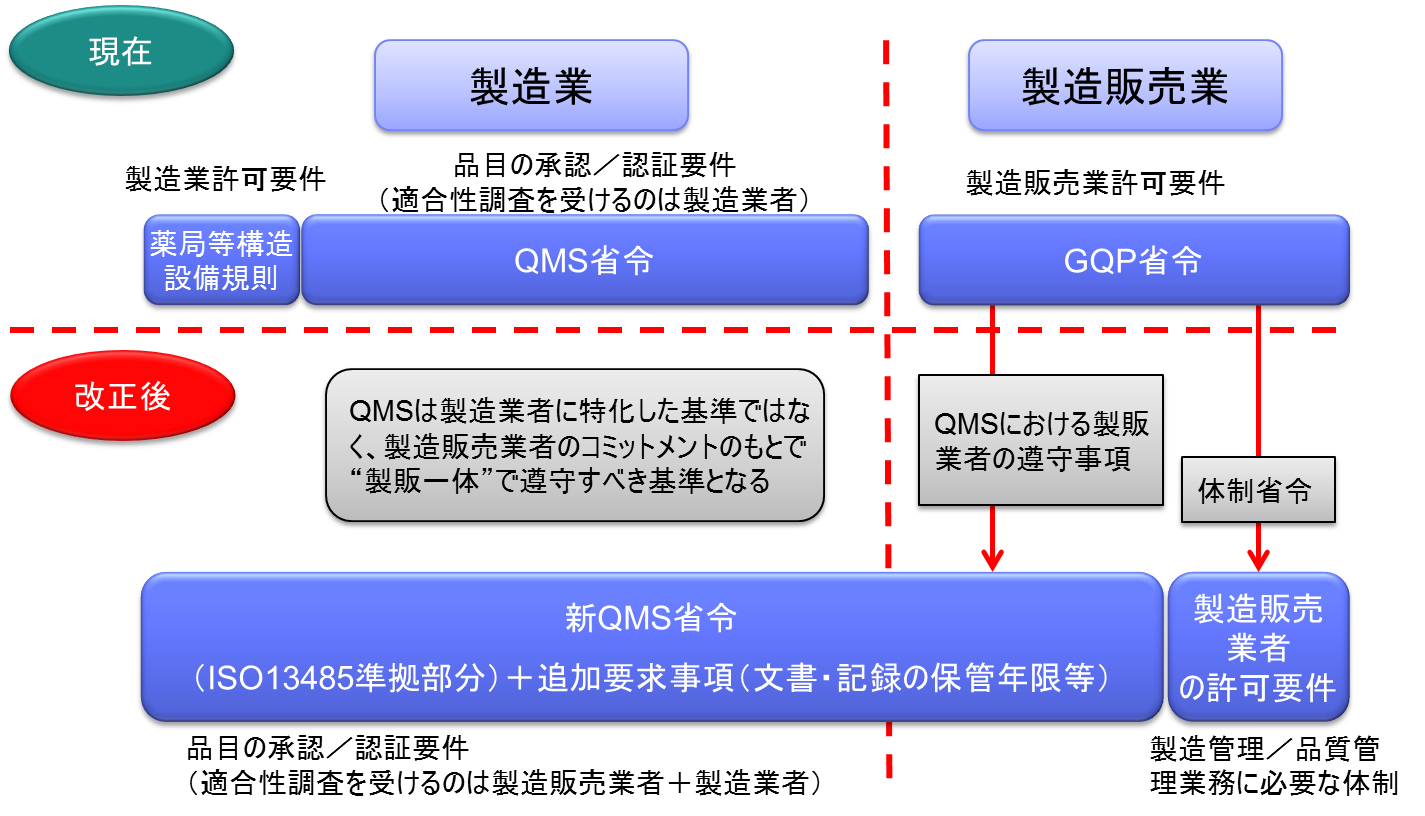
医薬品医療機器等法施行後は、製造販売業がQMS省令の対象となる。
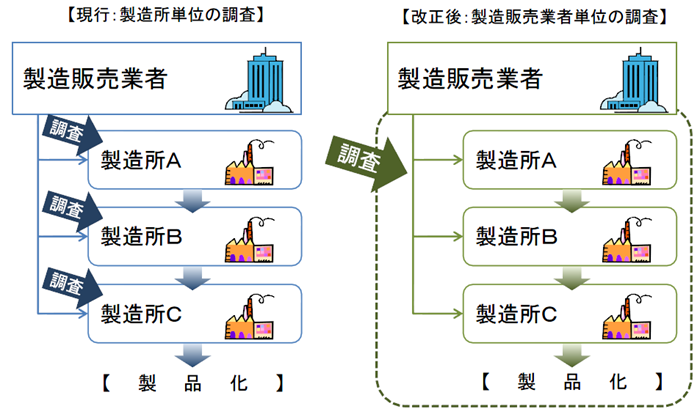
従来のように製造所毎に別個に調査・判定をするのではなく、製造販売業者に対して、品質システム全体を包括的に調査・判定することになった。
製造販売業者の責任業務
製造販売業者は、次に掲げる業務を行わなければならない。
- QMSに必要な工程の内容(当該工程により達成される結果を含む。)を明らかにするとともに、当該工程のそれぞれについて、各施設の関与の態様を明確にすること。
- 工程の順序および相互の関係を明確にすること。
- 工程の実施および管理の実効性の確保に必要な判定基準および方法を明確にすること。
- 工程の実施、監視および測定に必要な資源および情報が利用できるようにすること。
- 工程を監視し、測定し、および分析すること。
- 工程について、1.の結果を得るために、および実効性を維持するために所要の措置を採ること。
QMSの構築
製造販売業者は、新QMS省令にもとづき、QMSの構築を行わなければならない。
QMS適合性調査
医薬品医療機器等法施行後のQMS適合性調査は、製造販売業のQMSのもと、PMDAまたは登録認証機関が調査を実施する。(適合性調査を受けるのは製造販売業者+製造業者)
*QMS=企業がシステム(組織体制やルール)を確立し、製造に関わる組織全体で品質保証(製造管理及び品質管理)すること。
体制の整備
製造販売業許可を取得する場合、管理監督者(経営者)、管理責任者の他に、いわゆる三役(医療機器等総括製造販売責任者、国内品質業務運営責任者、医療機器等安全管理責任者)を置かなければならない。
また製造業の登録申請を行う場合、責任技術者を置かなければならない。
管理責任者
管理責任者は、製造販売業者等の役員、管理職の地位にある者その他これに相当する者がその任につくこと。
管理責任者の責任及び権限
- 工程が確立され、実施されるとともに、その実効性が維持されているようにすること。
- 品質管理監督システムの実施状況及びその改善の必要性について管理監督者に報告すること。
- 全ての施設において、法令の規定等及び製品受領者要求事項についての認識が向上するようにすること。
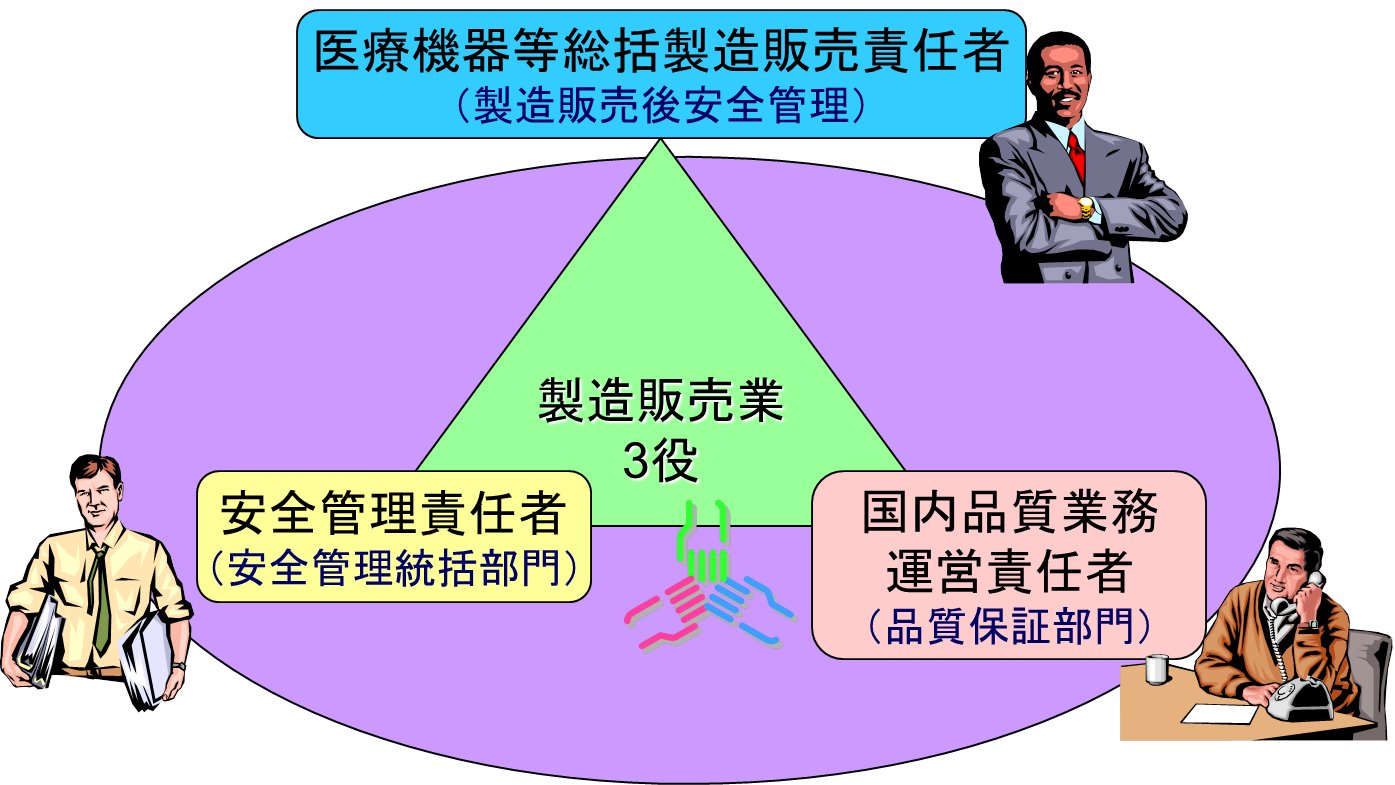
医療機器等総括製造販売責任者の業務
医療機器等総括製造販売責任者は、管理監督者、管理責任者、国内品質業務運営責任者を兼ねることができる。
- 製品の出荷の決定その他の製造管理及び品質管理に係る業務を統括し、これに責任を負うこと。
- 業務を公正かつ適正に行うために必要があると認めるときは、製造販売業者、管理監督者その他の当該業務に関して責任を有する者に対し文書により必要な意見を述べ、その写しを五年間保管すること。
- 国内品質業務運営責任者を監督すること。
- 管理責任者及び国内品質業務運営責任者(限定第三種医療機器製造販売業者にあっては、管理責任者を除く。)の意見を尊重すること。
- 安全管理統括部門との密接な連携を図らせること。
医療機器等総括製造販売責任者の要件
- クラス2以上(施行規則第114条の49第1項)
高度管理医療機器又は管理医療機器の製造管理及び品質管理並びに製造販売後安全管理を行う者に係る法第23条の2の14第1項の厚生労働省令で定める基準は、次の各号のいずれかに該当する者であること。
- 大学等で物理学、化学、生物学、工学、情報学、金属学、電気学、機械学、薬学、医学又は歯学に関する専門の課程を修了した者
- 旧制中学若しくは高校又はこれと同等以上の学校で、物理学、化学、生物学、工学、情報学、金属学、電気学、機械学、薬学、医学又は歯学に関する専門の課程を修了した後、医薬品、医療機器又は再生医療等製品の品質管理又は製造販売後安全管理に関する業務に3年以上従事した者
- 医薬品、医療機器又は再生医療等製品の品質管理又は製造販売後安全管理に関する業務に5年以上従事した後、別に厚生労働省令で定めるところにより厚生労働大臣の登録を受けた者が行う講習を修了した者
四 厚生労働大臣が前3号に掲げる者と同等以上の知識経験を有すると認めた者
- クラス1(規則第114条の49第2項)
一般医療機器の製造管理及び品質管理並びに製造販売後安全管理を行う者に係る法第23条の2の14第1項の厚生労働省令で定める基準は、次の各号のいずれかに該当する者であることとする。
- 旧制中学若しくは高校又はこれと同等以上の学校で、物理学、化学、生物学、工学、情報学、金属学、電気学、機械学、薬学、医学又は歯学に関する専門の課程を修了した者
- 旧制中学若しくは高校又はこれと同等以上の学校で、物理学、化学、生物学、工学、情報学、金属学、電気学、機械学、薬学、医学又は歯学に関する科目を修得した後、医薬品、医薬部外品、化粧品、医療機器又は再生医療等製品の品質管理又は製造販売後安全管理に関する業務に3年以上従事した者
- 厚生労働大臣が前2号に掲げる者と同等以上の知識経験を有すると認めた者
国内品質業務運営責任者の業務
国内品質業務運営責任者は、管理責任者を兼ねることができる。
- 国内の品質管理業務を統括すること。
- 国内の品質管理業務が適正かつ円滑に行われていることを確認すること。
- 国内に流通させる製品について、市場への出荷の決定をロット(製造番号又は製造記号)ごとに行い、その結果及び出荷先等市場への出荷の記録を作成すること。
- 国内に流通する製品について、当該製品の品質に影響を与えるおそれのある製造方法、試験検査方法等の変更がなされる場合にあっては、当該変更に係る情報を国内外から収集し、かつ、把握するとともに、当該変更が製品の品質に重大な影響を与えるおそれがある場合には、速やかに管理責任者及び医療機器等総括製造販売責任者に対して文書により報告し、必要かつ適切な措置が採られるようにすること。
- 国内に流通する製品について、当該製品の品質等に関する情報(品質不良又はそのおそれに係る情報を含む。)を国内外から収集するとともに、当該情報を得たときは、速やかに管理責任者及び医療機器等総括製造販売責任者に対して文書により報告し、記録し、及び必要かつ適切な措置が採られるようにすること。
- 国内に流通する製品の回収を行う場合に、次に掲げる業務を行うこと。
イ 回収した医療機器等を区分して一定期間保管した後、適正に処理すること。
ロ 回収の内容を記載した記録を作成し、管理責任者及び医療機器等総括製造販売責任者に対して文書により報告すること。 - 第4号から前号までに掲げるもののほか、国内の品質管理業務の遂行のために必要があると認めるときは、管理責任者及び医療機器等総括製造販売責任者に対して文書により報告すること。
- 国内の品質管理業務の実施に当たり、必要に応じ、関係する登録製造所に係る製造業者又は医療機器等外国製造業者、販売業者、薬局開設者、病院及び診療所の開設者その他関係者に対し、文書による連絡又は指示を行うこと。
- 製造販売後安全管理基準第二条第二項に規定する安全確保措置に関する情報を知ったときは、安全管理統括部門に遅滞なく文書で提供すること。
国内品質業務運営責任者の要件
製造販売業者は、この省令の規定に従って行う国内の製品の品質を管理する業務の責任者として、国内に所在する施設に、次に掲げる要件を満たす国内品質業務運営責任者を置かなければならない。
- 製造販売業者における品質保証部門の責任者であること。
- 品質管理業務その他これに類する業務に三年以上従事した者であること。
- 国内の品質管理業務を適正かつ円滑に遂行しうる能力を有する者であること。
- 医療機器等の販売に係る部門に属する者でないことその他国内の品質管理業務の適正かつ円滑な遂行に支障を及ぼすおそれがない者であること。
注)国内品質業務運営責任者は、従前のQMS省令では、品質保証責任者と呼んでいた。改正法では、医薬品と医療機器を区別したため、医療機器における名称が変更になった。企業は、今後ともSOPなどで品質保証責任者と呼んでも構わない。
安全管理責任者
第一種製造販売業者は、次に掲げる要件を満たす安全確保業務の統括に係る部門(以下この章において「安全管理統括部門」という。)を置かなければならない。
- 総括製造販売責任者の監督下にあること。
- 安全確保業務を適正かつ円滑に遂行しうる能力を有する人員を十分に有すること。
- 医薬品等の販売に係る部門その他安全確保業務の適正かつ円滑な遂行に支障を及ぼすおそれのある部門から独立していること。
第一種製造販売業者は、次に掲げる要件を満たす安全確保業務の責任者(以下この章において「安全管理責任者」という。)を置かなければならない。
- 安全管理統括部門の責任者であること。
- 安全確保業務その他これに類する業務に三年以上従事した者であること。
- 安全確保業務を適正かつ円滑に遂行しうる能力を有する者であること。
- 医薬品等の販売に係る部門に属する者でないことその他安全確保業務の適正かつ円滑な遂行に支障を及ぼすおそれがない者であること。
- 第一種製造販売業者は、次項に規定する場合を除き、安全管理責任者に安全確保業務を行わせなければならない。
- 第一種製造販売業者は、安全確保業務であって規則第九十七条 各号に掲げるものの全部又は一部を安全管理責任者以外の者に行わせる場合にあっては、当該業務を適正かつ円滑に遂行しうる能力を有する当該業務の実施に係る責任者(以下「安全管理実施責任者」という。)を置かなければならない。
プログラムに係る総括販売責任者等の資格要件に関する経過措置
プログラム医療機器のみを製造販売する製造販売業者の総括製造販売責任者、国内品質業務運営責任者若しくは安全管理責任者並びにプログラム医療機器のみを製造する製造所の医療機器責任技術者に係る基準について、平成29 年11 月24 日までの間は、プログラム医療機器に係る特別講習を修了した者を、3年以上の業務経験がある者とみなす。
製造業
医療機器とされるソフトウェア(例:診断用ソフトウェア)を設計する業者も製造業の登録が必要である。
経過措置として、登録対象製造所ごとに、施行日から起算して3か月以内(2014年11月25日~2015年2月24日)に登録の申請をしなければならない。
医療機器プログラム(単体プログラム)製造業の登録範囲
製造業については、登録対象となる範囲は、医療機器プログラム(医療機器のうちプログラムであるものをいう。以下同じ。)の場合は設計のみ、医療機器プログラムを記録した記録媒体の場合は設計及び国内における最終製品の保管となる。(新法第23 条の2の3、新施行規則第114 条の8関係)
製造業の登録申請
医療機器(ソフトウェアを含む)を製造する場合は、製造業として登録の申請(医薬品医療機器等法第二十三条の二の三第一項)を行う必要がある。登録の有効期限は5年間である。
ソフトウェアの設計を行う者も製造業の登録が必要であるため注意が必要である。
登録制への移行に伴い、登録申請時に添付する資料が簡素化された。
医療機器とされるソフトウェア(例:診断用ソフトウェア)を設計・開発・製造する業者も製造業の登録が必要である。経過措置として、登録対象製造所ごとに、施行日から起算して3か月以内に登録の申請をしなければならない。
すでに製造業許可を受けている製造所は、自動的に登録されるため、許可期限の残日数は何もしなくてもよい。許可更新予定日までに新たに登録することとなる。
【現行】すべて許可制 【改正後】医療機器、体外診断用医薬品は登録制へ
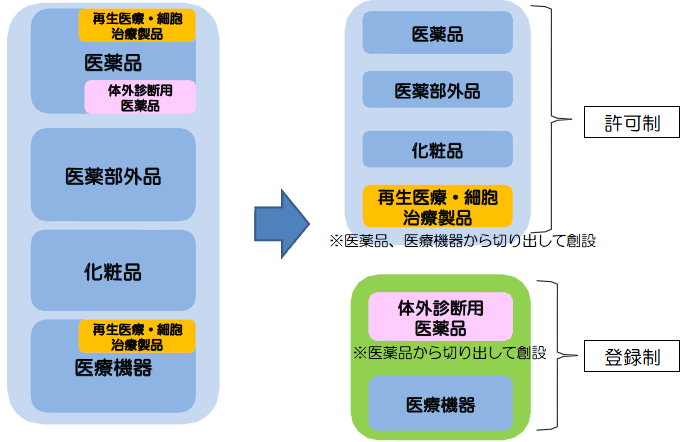
従来、許可申請時には所管都道府県によるQMS適合性調査が実施されていたが、医薬品医療機器等法施行後は、登録時に調査が行われることはない。
医療機器プログラムに関する表示
医療機器プログラムを記録した記録媒体またはその直接の容器もしくは被包への表示と電磁的記録による表示の両方が必要。
欧米で医療機器となるソフトウェアを販売するためには
一方で、海外においては、これまでも医療機器に搭載するソフトウェアの開発には、非常に厳格な規制要件の遵守が義務付けられてきた。
IEC-62304「Medical device software —Software life cycle processes」やFDAガイダンス「General Principles of Software Validation」等に対応しなければ海外展開できない。
ソフトウェアを搭載した医療機器の海外展開に際しては、IEC-62304に準拠してソフトウェアを設計開発することが必須である。
つまり、医療機器企業は、IEC-62304に準拠したソフトウェアの開発プロセスを構築しなければならない。
IEC-62304には、ライフサイクルプロセスの規格が概説され、ソフトウェア品質を確保するための製造者のアクティビティについて規定されている。
しかしながら、これまで国内向け医療機器製品は、IEC-62304などの海外規格を意識せずに開発をしていることが多いと思われる。
なぜならば、日本においては、これまでは医療機器ソフトウェア開発に関する規制がなかったからだ。
今後IEC-62304は、IEC 82304、IEC 80001の発行と相まって、改訂が予定されている。
米国FDAの規制要件
米国では、1985年から1987年にかけて、放射線治療装置のソフトウェアのバグにより、6名の犠牲者が出るといった事故が発生した。
この事故を受けて、FDAは1987年に『General Principles of Software Validation』(GPSV)を発行した。
米国に輸出するためには、GPSVにも準拠しなければならない。
IEC-62304の適合認証を受けていたにもかかわらず、FDAから厳しい指摘を受けることもある。
General Principles of Software Validationでは「医療機器ソフトウェアの妥当性確認または医療機器の設計、開発、製造に使用されるソフトウェアの妥当性確認に適用可能であると食品医薬品局(FDA)が考える、バリデーションに関する一般原則」の概要を示している。
これらの原則は以下のソフトウェアに適用される。
- 医療機器のコンポーネント、パーツ、又はアクセサリーとして用いられるソフトウェア
- 医療機器であるソフトウェア(例:血液組織ソフトウェア)
- 装置の製造に用いられるソフトウェア(例:製造機器内のPLC)
- 機器製造業者用品質システムの履行に用いられるソフトウェア(例:機器の履歴を記録、メンテナンスするソフトウェア)
ただし、FDAは特定の開発・バリデーションに関する手法には言及していない。
FDAによる主なガイダンスとして、
- Guidance for the Submission Of Pre-market Notifications for Medical Image Management Devices(医療画像処理機器の市販前通知申請に関するガイダンス)
- Guidance for the Content of Pre-market Submissions for Software Contained in Medical Devices(医療機器に含まれるソフトウェアの市販前申請に関するガイダンス)
- General Principles of Software Validation; Final Guidance for Industry and FDA Staff(ソフトウェアバリデーションの一般原則)
- Off-The-Shelf Software Use in Medical Devices(医療機器における市販ソフトを利用)
などがあり、また規制要件や業界標準は常に更新されるため、必要な規則を全て遵守することは容易ではない。
ソフトウェアのリスクマネジメント
また、製造者はISO 14971に適合するリスク管理プロセスを適用しなければならない。
リスク管理プロセスは複雑で難解である。
一般にソフトウェアのリスク分析では、FMEAを使用する。
しかしながら、具体的な実施方法がわからない場合がほとんどであろう。






