ISO-13485(QMS省令)とFDA QSRの違いについて ㈱イーコンプライアンス
FDA QSRとは
1996年10月7日、FDAは、1978年の医療機器GMP規則を改正して「品質システム(QS:Quality System)規則」と呼ばれる規則を公示した。
『21 CFR Part 820 Quality System Regulation - QSR』
QSRは、ISO 13485:1996/ISO 9001:1994を基礎にして作成された。
「品質システム規則」(QSR)は、医療機器の設計、購入、製造、包装、表示、保管、設置およびサービスで用いられる手順とこれらに対して用いられる設備および管理に関する要件を含む。
規則の定義では、品質(Quality)は安全性や性能を含めて使用適合性(Fitness-for-use)を満たすため機器の能力を支える特徴(Feature)と特質(Characteristic)の総体をいい、QSは品質管理の実施に対する組織構造、責任、手順、プロセスおよび資源をいう。
品質システム規則の一般規定は、次のようなことを定める。
- 各製造業者は設計または製造される特定の医療機器に対して適切な、そしてこの規定の要件に適合する品質システムを確立し、維持しなければならない。
- 規則に定める要件は最終機器が安全かつ有効であり、またFFDCAの遵守を保証する意図をもつ。
- 規則は最終機器の構成物または部品の製造業者には適用されないが、このような製造業者に対してガイダンスとしてこの規則の規定を用いることを勧める。
外国の製造業者については、アメリカに輸入される医療機器の外国製造業者が施設へのFDA査察を拒否する場合、その施設で製造される機器は法的に不良品(Adulterated product)とみなされる。
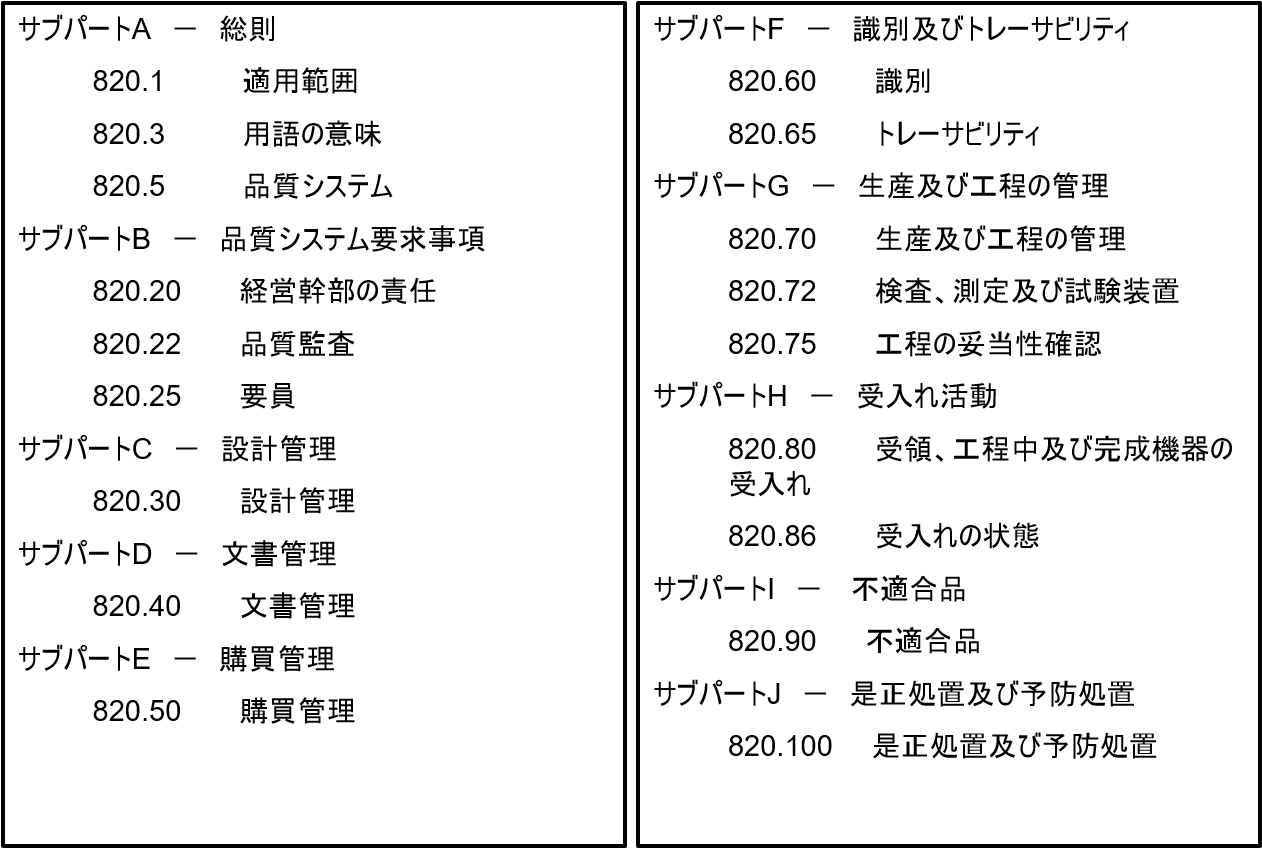
規則は、一般規定(範囲、定義、品質システム)、品質システム要件(管理責任、品質査察、従業員)、設計管理、文書管理、購入管理、確認と追跡(確認、追跡)、生産および工程管理(生産および工程管理、検査・測定・検査器具、工程バリデーション)、容認作業(受領、工程中および最終機器の容認、容認の状態)、不適合製品、是正および予防措置、表示および包装管理(機器表示、機器包装)、取扱・保管・流通・設置(取扱、保管、流通、設置)、記録(一般要件、機器マスター記録、機器歴記録、苦情ファイル)、サービング、統計技術の各項目(Subpart)を含む。
QSRの適用対象は、一部の例外を除く全クラスの医療機器である。
QSRは、クラスⅠ機器にも適用されるが、わずかに例外がある。例えば、顔弓、ラバーダム、味覚計、採尿器、弾性包帯は、QSR免除クラスⅠ機器である。
設計管理は、一部の例外を除き、クラスⅠ機器には適用されない。例外として、コンピュータソフトウェア自動化クラスⅠ機器などがある。
標題・・・Quality System Regulation: QSR(品質システム規則)
適用・・・人体用に意図された全ての医用完成機器の設計・購買・製造・包装・ラベリング・保管・取り付けおよび付帯サービス:医療機器の製造に関する基本的な要求事項
構成・・・サブパートAからOまでの15セクションに分かれている。
主な要件
- 品質システム要求事項
- 設計管理
- 文書管理
- 識別およびトレーサビリティ
- 生産および工程管理
- 是正処置および予防処置

ISO-13485(QMS省令)とFDA QSRの違いについて
QSRは、ISO 13485:1996/ISO 9001:1994を基礎にして法制化された。そのため、整合化された各国の規則と共通する要素が多く、実質的には同等とも言える。
ただし、FDA固有の要求事項が存在する。
- 設計管理において、リスクマネジメントではなく、リスク分析を要求
- 妥当性確認されたプロセスに対する監視及び管理の方法、要員、装置などの文書化を要求
- 機器原簿(Device Master Record - DMR)及び機器履歴簿(Device History Record - DHR)、品質システム記録(Quality System Record - QSR)、設計履歴ファイル(Design History File - DHF)の内容及び維持に関する要求
- 苦情ファイル(Complaint File)に関する詳細な要求など







